
Dear Freesurfer experts
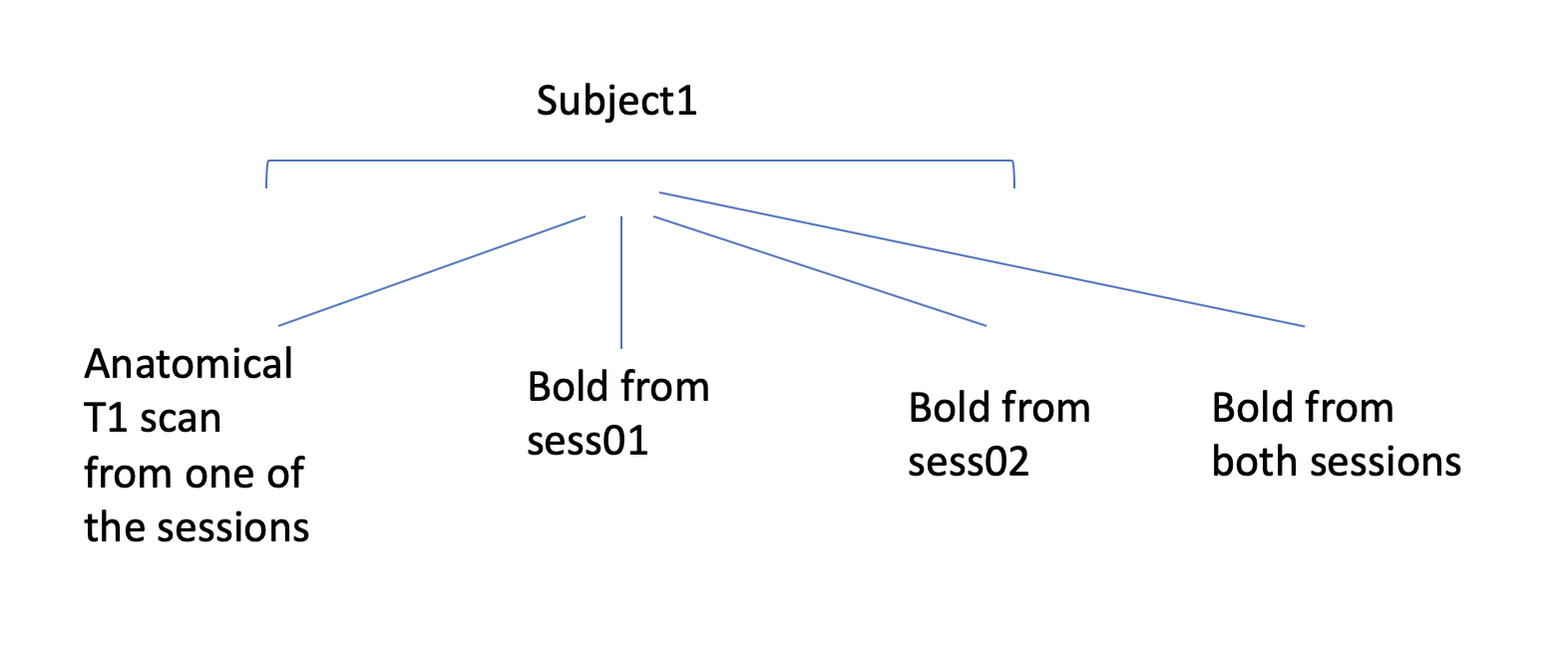
Happy Holidays! I have a subject with two highres fMRI scan sessions. Attached you can see an image, showing how I organized the sub-folders. I preprocessed the bold1 and bold2 and then copied the preprocessed files to "bold from both sessions" folder. I ran selxavg for bold1 and bold2 seperately and it went well. However, when I ran selxavg for "bold from both sessions" folder, it gives the below error. Could you please let me know what the problem is?Also if you have a better suggestion for how I should organize my subfolders/analysis, I appreciate that.
Thanks Mona
>> /autofs/cluster/freesurfer/centos7_x86_64/stable6/fsfast/toolbox/fast_selxavg3.m
/autofs/cluster/freesurfer/centos7_x86_64/stable6/fsfast/toolbox/fast_ldanaflac.m /autofs/cluster/freesurfer/centos7_x86_64/stable6/matlab/MRIread.m
starting fast_selxavg3b
#@# arap_7T_all ############################### /autofs/space/oprah_001/users/zn025/looming_7T/all_subjects/arap_7T_all ------------------------- $Id: fast_selxavg3b.m,v 1.4 2016/05/04 22:16:47 greve Exp $ ------------------------- outtop = /autofs/space/oprah_001/users/zn025/looming_7T/all_subjects Extension format = nii.gz INFO: key nSliceGroups unrecognized, line 12, skipping 1 app.mat 2 aw.mat 3 caw.mat 4 clo.mat 5 faw.mat 6 fem.mat 7 fm.mat 8 fmaw.mat 9 mal.mat 10 maw.mat 11 oaw.mat 12 oc.mat 13 ocaw.mat 14 ope.mat 15 wit.mat Excluding 6 points nruns = 15 autostimdur =
outanadir = /autofs/space/oprah_001/users/zn025/looming_7T/all_subjects/arap_7T_all/loom/loom_native.lh Excluding 6 points Excluding 0 points Excluding 1 points Excluding 5 points Excluding 8 points Excluding 3 points Excluding 8 points Excluding 4 points Excluding 6 points Excluding 10 points Excluding 5 points Excluding 5 points Excluding 5 points Excluding 5 points Excluding 5 points parfiles condition id list: 1 2 3 4 5 6 7 8 Found 41883/148565 (28.2) voxels in mask 1 Creating Design Matrix ... creation time = 0.040 sec DoMCFit = 1 ntptot = 1350, nX = 219, DOF = 1131 Saving X matrix to /autofs/space/oprah_001/users/zn025/looming_7T/all_subjects/arap_7T_all/loom/loom_native.lh/Xtmp.mat XCond = 1873.07 (normalized) Computing compensation for resdual AR1 bias 1 -0.5 -0.456169 (t=1.17104) 2 -0.25 -0.261165 (t=2.14097) 3 0 -0.076309 (t=2.3701) 4 0.25 0.0914527 (t=3.32529) 5 0.5 0.225506 (t=4.09175) AR1 Correction M: 0.138222 1.44983 Computing contrast matrices Computing contrasts Loading previous GLM fit Starting contrasts app J=1 ------------- Error using * Incorrect dimensions for matrix multiplication. Check that the number of columns in the first matrix matches the number of rows in the second matrix. To perform element wise multiplication, use '.*'.
Error in fast_fratiow (line 81) ces = C*beta;
Error in fast_selxavg3b (line 1153) fast_fratiow(betamat,X,rvarmat,C,acfsegmn,acfseg.vol(:));
ERROR: fast_selxavg3() failed\n
Zahra (Mona) Nasiriavanaki
Postdoctoral Research Fellow
Martinos Center for Biomedical Imaging
Massachusetts General Hospital
149 13th Street, 149-2615
Charlestown, MA, USA, 02129
{kind=link}

Why are you combining them into one bold folder?
On 12/30/2019 9:09 AM, Nasiriavanaki, Zahra wrote: Dear Freesurfer experts
Happy Holidays! I have a subject with two highres fMRI scan sessions. Attached you can see an image, showing how I organized the sub-folders. I preprocessed the bold1 and bold2 and then copied the preprocessed files to "bold from both sessions" folder. I ran selxavg for bold1 and bold2 seperately and it went well. However, when I ran selxavg for "bold from both sessions" folder, it gives the below error. Could you please let me know what the problem is?Also if you have a better suggestion for how I should organize my subfolders/analysis, I appreciate that.
Thanks Mona
>> /autofs/cluster/freesurfer/centos7_x86_64/stable6/fsfast/toolbox/fast_selxavg3.m
/autofs/cluster/freesurfer/centos7_x86_64/stable6/fsfast/toolbox/fast_ldanaflac.m /autofs/cluster/freesurfer/centos7_x86_64/stable6/matlab/MRIread.m
starting fast_selxavg3b
#@# arap_7T_all ############################### /autofs/space/oprah_001/users/zn025/looming_7T/all_subjects/arap_7T_all ------------------------- $Id: fast_selxavg3b.m,v 1.4 2016/05/04 22:16:47 greve Exp $ ------------------------- outtop = /autofs/space/oprah_001/users/zn025/looming_7T/all_subjects Extension format = nii.gz INFO: key nSliceGroups unrecognized, line 12, skipping 1 app.mat 2 aw.mat 3 caw.mat 4 clo.mat 5 faw.mat 6 fem.mat 7 fm.mat 8 fmaw.mat 9 mal.mat 10 maw.mat 11 oaw.mat 12 oc.mat 13 ocaw.mat 14 ope.mat 15 wit.mat Excluding 6 points nruns = 15 autostimdur =
outanadir = /autofs/space/oprah_001/users/zn025/looming_7T/all_subjects/arap_7T_all/loom/loom_native.lh Excluding 6 points Excluding 0 points Excluding 1 points Excluding 5 points Excluding 8 points Excluding 3 points Excluding 8 points Excluding 4 points Excluding 6 points Excluding 10 points Excluding 5 points Excluding 5 points Excluding 5 points Excluding 5 points Excluding 5 points parfiles condition id list: 1 2 3 4 5 6 7 8 Found 41883/148565 (28.2) voxels in mask 1 Creating Design Matrix ... creation time = 0.040 sec DoMCFit = 1 ntptot = 1350, nX = 219, DOF = 1131 Saving X matrix to /autofs/space/oprah_001/users/zn025/looming_7T/all_subjects/arap_7T_all/loom/loom_native.lh/Xtmp.mat XCond = 1873.07 (normalized) Computing compensation for resdual AR1 bias 1 -0.5 -0.456169 (t=1.17104) 2 -0.25 -0.261165 (t=2.14097) 3 0 -0.076309 (t=2.3701) 4 0.25 0.0914527 (t=3.32529) 5 0.5 0.225506 (t=4.09175) AR1 Correction M: 0.138222 1.44983 Computing contrast matrices Computing contrasts Loading previous GLM fit Starting contrasts app J=1 ------------- Error using * Incorrect dimensions for matrix multiplication. Check that the number of columns in the first matrix matches the number of rows in the second matrix. To perform element wise multiplication, use '.*'.
Error in fast_fratiow (line 81) ces = C*beta;
Error in fast_selxavg3b (line 1153) fast_fratiow(betamat,X,rvarmat,C,acfsegmn,acfseg.vol(:));
ERROR: fast_selxavg3() failed\n
Zahra (Mona) Nasiriavanaki
Postdoctoral Research Fellow
Martinos Center for Biomedical Imaging
Massachusetts General Hospital
149 13th Street, 149-2615
Charlestown, MA, USA, 02129
_______________________________________________ Freesurfer mailing list Freesurfer@nmr.mgh.harvard.edumailto:Freesurfer@nmr.mgh.harvard.edu https://mail.nmr.mgh.harvard.edu/mailman/listinfo/freesurfer
I need to concatenate the two sessions. We scanned the subjects twice with the same task. I need to see the maps for each session separately and also together.
Thanks
Zahra (Mona) Nasiriavanaki
Postdoctoral Research Fellow
Martinos Center for Biomedical Imaging
Massachusetts General Hospital
149 13th Street, 149-2615
Charlestown, MA, USA, 02129
________________________________ From: freesurfer-bounces@nmr.mgh.harvard.edu freesurfer-bounces@nmr.mgh.harvard.edu on behalf of Greve, Douglas N.,Ph.D. DGREVE@mgh.harvard.edu Sent: Thursday, January 2, 2020 10:57 AM To: freesurfer@nmr.mgh.harvard.edu freesurfer@nmr.mgh.harvard.edu Subject: Re: [Freesurfer] highres fMRI selxavg error
Why are you combining them into one bold folder?
On 12/30/2019 9:09 AM, Nasiriavanaki, Zahra wrote: Dear Freesurfer experts
Happy Holidays! I have a subject with two highres fMRI scan sessions. Attached you can see an image, showing how I organized the sub-folders. I preprocessed the bold1 and bold2 and then copied the preprocessed files to "bold from both sessions" folder. I ran selxavg for bold1 and bold2 seperately and it went well. However, when I ran selxavg for "bold from both sessions" folder, it gives the below error. Could you please let me know what the problem is?Also if you have a better suggestion for how I should organize my subfolders/analysis, I appreciate that.
Thanks Mona
>> /autofs/cluster/freesurfer/centos7_x86_64/stable6/fsfast/toolbox/fast_selxavg3.m
/autofs/cluster/freesurfer/centos7_x86_64/stable6/fsfast/toolbox/fast_ldanaflac.m /autofs/cluster/freesurfer/centos7_x86_64/stable6/matlab/MRIread.m
starting fast_selxavg3b
#@# arap_7T_all ############################### /autofs/space/oprah_001/users/zn025/looming_7T/all_subjects/arap_7T_all ------------------------- $Id: fast_selxavg3b.m,v 1.4 2016/05/04 22:16:47 greve Exp $ ------------------------- outtop = /autofs/space/oprah_001/users/zn025/looming_7T/all_subjects Extension format = nii.gz INFO: key nSliceGroups unrecognized, line 12, skipping 1 app.mat 2 aw.mat 3 caw.mat 4 clo.mat 5 faw.mat 6 fem.mat 7 fm.mat 8 fmaw.mat 9 mal.mat 10 maw.mat 11 oaw.mat 12 oc.mat 13 ocaw.mat 14 ope.mat 15 wit.mat Excluding 6 points nruns = 15 autostimdur =
outanadir = /autofs/space/oprah_001/users/zn025/looming_7T/all_subjects/arap_7T_all/loom/loom_native.lh Excluding 6 points Excluding 0 points Excluding 1 points Excluding 5 points Excluding 8 points Excluding 3 points Excluding 8 points Excluding 4 points Excluding 6 points Excluding 10 points Excluding 5 points Excluding 5 points Excluding 5 points Excluding 5 points Excluding 5 points parfiles condition id list: 1 2 3 4 5 6 7 8 Found 41883/148565 (28.2) voxels in mask 1 Creating Design Matrix ... creation time = 0.040 sec DoMCFit = 1 ntptot = 1350, nX = 219, DOF = 1131 Saving X matrix to /autofs/space/oprah_001/users/zn025/looming_7T/all_subjects/arap_7T_all/loom/loom_native.lh/Xtmp.mat XCond = 1873.07 (normalized) Computing compensation for resdual AR1 bias 1 -0.5 -0.456169 (t=1.17104) 2 -0.25 -0.261165 (t=2.14097) 3 0 -0.076309 (t=2.3701) 4 0.25 0.0914527 (t=3.32529) 5 0.5 0.225506 (t=4.09175) AR1 Correction M: 0.138222 1.44983 Computing contrast matrices Computing contrasts Loading previous GLM fit Starting contrasts app J=1 ------------- Error using * Incorrect dimensions for matrix multiplication. Check that the number of columns in the first matrix matches the number of rows in the second matrix. To perform element wise multiplication, use '.*'.
Error in fast_fratiow (line 81) ces = C*beta;
Error in fast_selxavg3b (line 1153) fast_fratiow(betamat,X,rvarmat,C,acfsegmn,acfseg.vol(:));
ERROR: fast_selxavg3() failed\n
Zahra (Mona) Nasiriavanaki
Postdoctoral Research Fellow
Martinos Center for Biomedical Imaging
Massachusetts General Hospital
149 13th Street, 149-2615
Charlestown, MA, USA, 02129
_______________________________________________ Freesurfer mailing list Freesurfer@nmr.mgh.harvard.edumailto:Freesurfer@nmr.mgh.harvard.edu https://mail.nmr.mgh.harvard.edu/mailman/listinfo/freesurfer
When you say you need to see them together, what does that mean? Are you computing contrasts between the two sessions or you just want to combine them statistically?
On 1/2/2020 10:07 AM, Nasiriavanaki, Zahra wrote: I need to concatenate the two sessions. We scanned the subjects twice with the same task. I need to see the maps for each session separately and also together.
Thanks
Zahra (Mona) Nasiriavanaki
Postdoctoral Research Fellow
Martinos Center for Biomedical Imaging
Massachusetts General Hospital
149 13th Street, 149-2615
Charlestown, MA, USA, 02129
________________________________ From: freesurfer-bounces@nmr.mgh.harvard.edumailto:freesurfer-bounces@nmr.mgh.harvard.edu freesurfer-bounces@nmr.mgh.harvard.edumailto:freesurfer-bounces@nmr.mgh.harvard.edu on behalf of Greve, Douglas N.,Ph.D. DGREVE@mgh.harvard.edumailto:DGREVE@mgh.harvard.edu Sent: Thursday, January 2, 2020 10:57 AM To: freesurfer@nmr.mgh.harvard.edumailto:freesurfer@nmr.mgh.harvard.edu freesurfer@nmr.mgh.harvard.edumailto:freesurfer@nmr.mgh.harvard.edu Subject: Re: [Freesurfer] highres fMRI selxavg error
Why are you combining them into one bold folder?
On 12/30/2019 9:09 AM, Nasiriavanaki, Zahra wrote: Dear Freesurfer experts
Happy Holidays! I have a subject with two highres fMRI scan sessions. Attached you can see an image, showing how I organized the sub-folders. I preprocessed the bold1 and bold2 and then copied the preprocessed files to "bold from both sessions" folder. I ran selxavg for bold1 and bold2 seperately and it went well. However, when I ran selxavg for "bold from both sessions" folder, it gives the below error. Could you please let me know what the problem is?Also if you have a better suggestion for how I should organize my subfolders/analysis, I appreciate that.
Thanks Mona
>> /autofs/cluster/freesurfer/centos7_x86_64/stable6/fsfast/toolbox/fast_selxavg3.m
/autofs/cluster/freesurfer/centos7_x86_64/stable6/fsfast/toolbox/fast_ldanaflac.m /autofs/cluster/freesurfer/centos7_x86_64/stable6/matlab/MRIread.m
starting fast_selxavg3b
#@# arap_7T_all ############################### /autofs/space/oprah_001/users/zn025/looming_7T/all_subjects/arap_7T_all ------------------------- $Id: fast_selxavg3b.m,v 1.4 2016/05/04 22:16:47 greve Exp $ ------------------------- outtop = /autofs/space/oprah_001/users/zn025/looming_7T/all_subjects Extension format = nii.gz INFO: key nSliceGroups unrecognized, line 12, skipping 1 app.mat 2 aw.mat 3 caw.mat 4 clo.mat 5 faw.mat 6 fem.mat 7 fm.mat 8 fmaw.mat 9 mal.mat 10 maw.mat 11 oaw.mat 12 oc.mat 13 ocaw.mat 14 ope.mat 15 wit.mat Excluding 6 points nruns = 15 autostimdur =
outanadir = /autofs/space/oprah_001/users/zn025/looming_7T/all_subjects/arap_7T_all/loom/loom_native.lh Excluding 6 points Excluding 0 points Excluding 1 points Excluding 5 points Excluding 8 points Excluding 3 points Excluding 8 points Excluding 4 points Excluding 6 points Excluding 10 points Excluding 5 points Excluding 5 points Excluding 5 points Excluding 5 points Excluding 5 points parfiles condition id list: 1 2 3 4 5 6 7 8 Found 41883/148565 (28.2) voxels in mask 1 Creating Design Matrix ... creation time = 0.040 sec DoMCFit = 1 ntptot = 1350, nX = 219, DOF = 1131 Saving X matrix to /autofs/space/oprah_001/users/zn025/looming_7T/all_subjects/arap_7T_all/loom/loom_native.lh/Xtmp.mat XCond = 1873.07 (normalized) Computing compensation for resdual AR1 bias 1 -0.5 -0.456169 (t=1.17104) 2 -0.25 -0.261165 (t=2.14097) 3 0 -0.076309 (t=2.3701) 4 0.25 0.0914527 (t=3.32529) 5 0.5 0.225506 (t=4.09175) AR1 Correction M: 0.138222 1.44983 Computing contrast matrices Computing contrasts Loading previous GLM fit Starting contrasts app J=1 ------------- Error using * Incorrect dimensions for matrix multiplication. Check that the number of columns in the first matrix matches the number of rows in the second matrix. To perform element wise multiplication, use '.*'.
Error in fast_fratiow (line 81) ces = C*beta;
Error in fast_selxavg3b (line 1153) fast_fratiow(betamat,X,rvarmat,C,acfsegmn,acfseg.vol(:));
ERROR: fast_selxavg3() failed\n
Zahra (Mona) Nasiriavanaki
Postdoctoral Research Fellow
Martinos Center for Biomedical Imaging
Massachusetts General Hospital
149 13th Street, 149-2615
Charlestown, MA, USA, 02129
_______________________________________________ Freesurfer mailing list Freesurfer@nmr.mgh.harvard.edumailto:Freesurfer@nmr.mgh.harvard.edu https://mail.nmr.mgh.harvard.edu/mailman/listinfo/freesurfer
I want to concatenate the two sessions which I guess is the same as combining them statistically. Please note that it's all done in "self" space not fsaverage.
Zahra (Mona) Nasiriavanaki
Postdoctoral Research Fellow
Martinos Center for Biomedical Imaging
Massachusetts General Hospital
149 13th Street, 149-2615
Charlestown, MA, USA, 02129
________________________________ From: Greve, Douglas N.,Ph.D. DGREVE@mgh.harvard.edu Sent: Thursday, January 2, 2020 11:46 AM To: Nasiriavanaki, Zahra ZNASIRIAVANAKI@mgh.harvard.edu; freesurfer@nmr.mgh.harvard.edu freesurfer@nmr.mgh.harvard.edu Subject: Re: [Freesurfer] highres fMRI selxavg error
When you say you need to see them together, what does that mean? Are you computing contrasts between the two sessions or you just want to combine them statistically?
On 1/2/2020 10:07 AM, Nasiriavanaki, Zahra wrote: I need to concatenate the two sessions. We scanned the subjects twice with the same task. I need to see the maps for each session separately and also together.
Thanks
Zahra (Mona) Nasiriavanaki
Postdoctoral Research Fellow
Martinos Center for Biomedical Imaging
Massachusetts General Hospital
149 13th Street, 149-2615
Charlestown, MA, USA, 02129
________________________________ From: freesurfer-bounces@nmr.mgh.harvard.edumailto:freesurfer-bounces@nmr.mgh.harvard.edu freesurfer-bounces@nmr.mgh.harvard.edumailto:freesurfer-bounces@nmr.mgh.harvard.edu on behalf of Greve, Douglas N.,Ph.D. DGREVE@mgh.harvard.edumailto:DGREVE@mgh.harvard.edu Sent: Thursday, January 2, 2020 10:57 AM To: freesurfer@nmr.mgh.harvard.edumailto:freesurfer@nmr.mgh.harvard.edu freesurfer@nmr.mgh.harvard.edumailto:freesurfer@nmr.mgh.harvard.edu Subject: Re: [Freesurfer] highres fMRI selxavg error
Why are you combining them into one bold folder?
On 12/30/2019 9:09 AM, Nasiriavanaki, Zahra wrote: Dear Freesurfer experts
Happy Holidays! I have a subject with two highres fMRI scan sessions. Attached you can see an image, showing how I organized the sub-folders. I preprocessed the bold1 and bold2 and then copied the preprocessed files to "bold from both sessions" folder. I ran selxavg for bold1 and bold2 seperately and it went well. However, when I ran selxavg for "bold from both sessions" folder, it gives the below error. Could you please let me know what the problem is?Also if you have a better suggestion for how I should organize my subfolders/analysis, I appreciate that.
Thanks Mona
>> /autofs/cluster/freesurfer/centos7_x86_64/stable6/fsfast/toolbox/fast_selxavg3.m
/autofs/cluster/freesurfer/centos7_x86_64/stable6/fsfast/toolbox/fast_ldanaflac.m /autofs/cluster/freesurfer/centos7_x86_64/stable6/matlab/MRIread.m
starting fast_selxavg3b
#@# arap_7T_all ############################### /autofs/space/oprah_001/users/zn025/looming_7T/all_subjects/arap_7T_all ------------------------- $Id: fast_selxavg3b.m,v 1.4 2016/05/04 22:16:47 greve Exp $ ------------------------- outtop = /autofs/space/oprah_001/users/zn025/looming_7T/all_subjects Extension format = nii.gz INFO: key nSliceGroups unrecognized, line 12, skipping 1 app.mat 2 aw.mat 3 caw.mat 4 clo.mat 5 faw.mat 6 fem.mat 7 fm.mat 8 fmaw.mat 9 mal.mat 10 maw.mat 11 oaw.mat 12 oc.mat 13 ocaw.mat 14 ope.mat 15 wit.mat Excluding 6 points nruns = 15 autostimdur =
outanadir = /autofs/space/oprah_001/users/zn025/looming_7T/all_subjects/arap_7T_all/loom/loom_native.lh Excluding 6 points Excluding 0 points Excluding 1 points Excluding 5 points Excluding 8 points Excluding 3 points Excluding 8 points Excluding 4 points Excluding 6 points Excluding 10 points Excluding 5 points Excluding 5 points Excluding 5 points Excluding 5 points Excluding 5 points parfiles condition id list: 1 2 3 4 5 6 7 8 Found 41883/148565 (28.2) voxels in mask 1 Creating Design Matrix ... creation time = 0.040 sec DoMCFit = 1 ntptot = 1350, nX = 219, DOF = 1131 Saving X matrix to /autofs/space/oprah_001/users/zn025/looming_7T/all_subjects/arap_7T_all/loom/loom_native.lh/Xtmp.mat XCond = 1873.07 (normalized) Computing compensation for resdual AR1 bias 1 -0.5 -0.456169 (t=1.17104) 2 -0.25 -0.261165 (t=2.14097) 3 0 -0.076309 (t=2.3701) 4 0.25 0.0914527 (t=3.32529) 5 0.5 0.225506 (t=4.09175) AR1 Correction M: 0.138222 1.44983 Computing contrast matrices Computing contrasts Loading previous GLM fit Starting contrasts app J=1 ------------- Error using * Incorrect dimensions for matrix multiplication. Check that the number of columns in the first matrix matches the number of rows in the second matrix. To perform element wise multiplication, use '.*'.
Error in fast_fratiow (line 81) ces = C*beta;
Error in fast_selxavg3b (line 1153) fast_fratiow(betamat,X,rvarmat,C,acfsegmn,acfseg.vol(:));
ERROR: fast_selxavg3() failed\n
Zahra (Mona) Nasiriavanaki
Postdoctoral Research Fellow
Martinos Center for Biomedical Imaging
Massachusetts General Hospital
149 13th Street, 149-2615
Charlestown, MA, USA, 02129
_______________________________________________ Freesurfer mailing list Freesurfer@nmr.mgh.harvard.edumailto:Freesurfer@nmr.mgh.harvard.edu https://mail.nmr.mgh.harvard.edu/mailman/listinfo/freesurfer
The easiest thing to do is to probably do the concatenation by hand, eg, mri_concat subj/sess01/analysis/contrast/ces.nii.gz subj/sess01/analysis/contrast/ces.nii.gz --o ces.sess01+sess02.nii.gz mri_concat subj/sess01/analysis/contrast/cesvar.nii.gz subj/sess01/analysis/contrast/cesvar.nii.gz --o cesvar.sess01+sess02.nii.gz
Create a dof file by adding together the dofs for each individual analysis as indicated by these values. Just put this number into a text file, eg, dof.sess01+sess02.dat cat subj/sess01/analysis/dof.dat subj/sess01/analysis/dof.dat
Create a mask fscalc subj/sess01/analysis/mask.nii.gz and subj/sess01/analysis/mask.nii.gz -o mask.sess01+sess02.nii.gz
Then run glmfit like mri_glmfit --surface subject lh --y ces.sess01+sess02.nii.gz --yffxvar cesvar.sess01+sess02.nii.gz --ffxdofdat dof.sess01+sess02.dat --mask mask.sess01+sess02.nii.gz --osgm --o ffx/subject/analysis/contrast/osgm
On 1/2/2020 11:20 AM, Nasiriavanaki, Zahra wrote: I want to concatenate the two sessions which I guess is the same as combining them statistically. Please note that it's all done in "self" space not fsaverage.
Zahra (Mona) Nasiriavanaki
Postdoctoral Research Fellow
Martinos Center for Biomedical Imaging
Massachusetts General Hospital
149 13th Street, 149-2615
Charlestown, MA, USA, 02129
________________________________ From: Greve, Douglas N.,Ph.D. DGREVE@mgh.harvard.edumailto:DGREVE@mgh.harvard.edu Sent: Thursday, January 2, 2020 11:46 AM To: Nasiriavanaki, Zahra ZNASIRIAVANAKI@mgh.harvard.edumailto:ZNASIRIAVANAKI@mgh.harvard.edu; freesurfer@nmr.mgh.harvard.edumailto:freesurfer@nmr.mgh.harvard.edu freesurfer@nmr.mgh.harvard.edumailto:freesurfer@nmr.mgh.harvard.edu Subject: Re: [Freesurfer] highres fMRI selxavg error
When you say you need to see them together, what does that mean? Are you computing contrasts between the two sessions or you just want to combine them statistically?
On 1/2/2020 10:07 AM, Nasiriavanaki, Zahra wrote: I need to concatenate the two sessions. We scanned the subjects twice with the same task. I need to see the maps for each session separately and also together.
Thanks
Zahra (Mona) Nasiriavanaki
Postdoctoral Research Fellow
Martinos Center for Biomedical Imaging
Massachusetts General Hospital
149 13th Street, 149-2615
Charlestown, MA, USA, 02129
________________________________ From: freesurfer-bounces@nmr.mgh.harvard.edumailto:freesurfer-bounces@nmr.mgh.harvard.edu freesurfer-bounces@nmr.mgh.harvard.edumailto:freesurfer-bounces@nmr.mgh.harvard.edu on behalf of Greve, Douglas N.,Ph.D. DGREVE@mgh.harvard.edumailto:DGREVE@mgh.harvard.edu Sent: Thursday, January 2, 2020 10:57 AM To: freesurfer@nmr.mgh.harvard.edumailto:freesurfer@nmr.mgh.harvard.edu freesurfer@nmr.mgh.harvard.edumailto:freesurfer@nmr.mgh.harvard.edu Subject: Re: [Freesurfer] highres fMRI selxavg error
Why are you combining them into one bold folder?
On 12/30/2019 9:09 AM, Nasiriavanaki, Zahra wrote: Dear Freesurfer experts
Happy Holidays! I have a subject with two highres fMRI scan sessions. Attached you can see an image, showing how I organized the sub-folders. I preprocessed the bold1 and bold2 and then copied the preprocessed files to "bold from both sessions" folder. I ran selxavg for bold1 and bold2 seperately and it went well. However, when I ran selxavg for "bold from both sessions" folder, it gives the below error. Could you please let me know what the problem is?Also if you have a better suggestion for how I should organize my subfolders/analysis, I appreciate that.
Thanks Mona
>> /autofs/cluster/freesurfer/centos7_x86_64/stable6/fsfast/toolbox/fast_selxavg3.m
/autofs/cluster/freesurfer/centos7_x86_64/stable6/fsfast/toolbox/fast_ldanaflac.m /autofs/cluster/freesurfer/centos7_x86_64/stable6/matlab/MRIread.m
starting fast_selxavg3b
#@# arap_7T_all ############################### /autofs/space/oprah_001/users/zn025/looming_7T/all_subjects/arap_7T_all ------------------------- $Id: fast_selxavg3b.m,v 1.4 2016/05/04 22:16:47 greve Exp $ ------------------------- outtop = /autofs/space/oprah_001/users/zn025/looming_7T/all_subjects Extension format = nii.gz INFO: key nSliceGroups unrecognized, line 12, skipping 1 app.mat 2 aw.mat 3 caw.mat 4 clo.mat 5 faw.mat 6 fem.mat 7 fm.mat 8 fmaw.mat 9 mal.mat 10 maw.mat 11 oaw.mat 12 oc.mat 13 ocaw.mat 14 ope.mat 15 wit.mat Excluding 6 points nruns = 15 autostimdur =
outanadir = /autofs/space/oprah_001/users/zn025/looming_7T/all_subjects/arap_7T_all/loom/loom_native.lh Excluding 6 points Excluding 0 points Excluding 1 points Excluding 5 points Excluding 8 points Excluding 3 points Excluding 8 points Excluding 4 points Excluding 6 points Excluding 10 points Excluding 5 points Excluding 5 points Excluding 5 points Excluding 5 points Excluding 5 points parfiles condition id list: 1 2 3 4 5 6 7 8 Found 41883/148565 (28.2) voxels in mask 1 Creating Design Matrix ... creation time = 0.040 sec DoMCFit = 1 ntptot = 1350, nX = 219, DOF = 1131 Saving X matrix to /autofs/space/oprah_001/users/zn025/looming_7T/all_subjects/arap_7T_all/loom/loom_native.lh/Xtmp.mat XCond = 1873.07 (normalized) Computing compensation for resdual AR1 bias 1 -0.5 -0.456169 (t=1.17104) 2 -0.25 -0.261165 (t=2.14097) 3 0 -0.076309 (t=2.3701) 4 0.25 0.0914527 (t=3.32529) 5 0.5 0.225506 (t=4.09175) AR1 Correction M: 0.138222 1.44983 Computing contrast matrices Computing contrasts Loading previous GLM fit Starting contrasts app J=1 ------------- Error using * Incorrect dimensions for matrix multiplication. Check that the number of columns in the first matrix matches the number of rows in the second matrix. To perform element wise multiplication, use '.*'.
Error in fast_fratiow (line 81) ces = C*beta;
Error in fast_selxavg3b (line 1153) fast_fratiow(betamat,X,rvarmat,C,acfsegmn,acfseg.vol(:));
ERROR: fast_selxavg3() failed\n
Zahra (Mona) Nasiriavanaki
Postdoctoral Research Fellow
Martinos Center for Biomedical Imaging
Massachusetts General Hospital
149 13th Street, 149-2615
Charlestown, MA, USA, 02129
_______________________________________________ Freesurfer mailing list Freesurfer@nmr.mgh.harvard.edumailto:Freesurfer@nmr.mgh.harvard.edu https://mail.nmr.mgh.harvard.edu/mailman/listinfo/freesurfer
Thanks a lot for your reply.
I have one more question about selxavg error and I appreciate if you could reply. I have another subject which has been scanned in 7T. When I run selxavg, it doesn't go through for the left and right hemi, but it gets perfectly done for subcortex. I attached the log file. We have collected the functional data in a slab of the brain containing subcortical areas and the insular cortex (at least this is what we aimed for). Looking at the data with tkregister2 command, I can't say if the insular cortex is included or not. Also, if it is included, then should we see the insular activation in cortical maps or subcortical ones? tkregister2 --s ylug_ass_sess01 --mov $all_subjects/ylug_ass_sess01/bold/010/template.nii.gz --surf --reg new.dat --regheader
Thanks a lot Mona
Zahra (Mona) Nasiriavanaki
Postdoctoral Research Fellow
Martinos Center for Biomedical Imaging
Massachusetts General Hospital
149 13th Street, 149-2615
Charlestown, MA, USA, 02129
________________________________ From: Greve, Douglas N.,Ph.D. DGREVE@mgh.harvard.edu Sent: Thursday, January 2, 2020 1:18 PM To: Nasiriavanaki, Zahra ZNASIRIAVANAKI@mgh.harvard.edu; freesurfer@nmr.mgh.harvard.edu freesurfer@nmr.mgh.harvard.edu Subject: Re: [Freesurfer] highres fMRI selxavg error
The easiest thing to do is to probably do the concatenation by hand, eg, mri_concat subj/sess01/analysis/contrast/ces.nii.gz subj/sess01/analysis/contrast/ces.nii.gz --o ces.sess01+sess02.nii.gz mri_concat subj/sess01/analysis/contrast/cesvar.nii.gz subj/sess01/analysis/contrast/cesvar.nii.gz --o cesvar.sess01+sess02.nii.gz
Create a dof file by adding together the dofs for each individual analysis as indicated by these values. Just put this number into a text file, eg, dof.sess01+sess02.dat cat subj/sess01/analysis/dof.dat subj/sess01/analysis/dof.dat
Create a mask fscalc subj/sess01/analysis/mask.nii.gz and subj/sess01/analysis/mask.nii.gz -o mask.sess01+sess02.nii.gz
Then run glmfit like mri_glmfit --surface subject lh --y ces.sess01+sess02.nii.gz --yffxvar cesvar.sess01+sess02.nii.gz --ffxdofdat dof.sess01+sess02.dat --mask mask.sess01+sess02.nii.gz --osgm --o ffx/subject/analysis/contrast/osgm
On 1/2/2020 11:20 AM, Nasiriavanaki, Zahra wrote: I want to concatenate the two sessions which I guess is the same as combining them statistically. Please note that it's all done in "self" space not fsaverage.
Zahra (Mona) Nasiriavanaki
Postdoctoral Research Fellow
Martinos Center for Biomedical Imaging
Massachusetts General Hospital
149 13th Street, 149-2615
Charlestown, MA, USA, 02129
________________________________ From: Greve, Douglas N.,Ph.D. DGREVE@mgh.harvard.edumailto:DGREVE@mgh.harvard.edu Sent: Thursday, January 2, 2020 11:46 AM To: Nasiriavanaki, Zahra ZNASIRIAVANAKI@mgh.harvard.edumailto:ZNASIRIAVANAKI@mgh.harvard.edu; freesurfer@nmr.mgh.harvard.edumailto:freesurfer@nmr.mgh.harvard.edu freesurfer@nmr.mgh.harvard.edumailto:freesurfer@nmr.mgh.harvard.edu Subject: Re: [Freesurfer] highres fMRI selxavg error
When you say you need to see them together, what does that mean? Are you computing contrasts between the two sessions or you just want to combine them statistically?
On 1/2/2020 10:07 AM, Nasiriavanaki, Zahra wrote: I need to concatenate the two sessions. We scanned the subjects twice with the same task. I need to see the maps for each session separately and also together.
Thanks
Zahra (Mona) Nasiriavanaki
Postdoctoral Research Fellow
Martinos Center for Biomedical Imaging
Massachusetts General Hospital
149 13th Street, 149-2615
Charlestown, MA, USA, 02129
________________________________ From: freesurfer-bounces@nmr.mgh.harvard.edumailto:freesurfer-bounces@nmr.mgh.harvard.edu freesurfer-bounces@nmr.mgh.harvard.edumailto:freesurfer-bounces@nmr.mgh.harvard.edu on behalf of Greve, Douglas N.,Ph.D. DGREVE@mgh.harvard.edumailto:DGREVE@mgh.harvard.edu Sent: Thursday, January 2, 2020 10:57 AM To: freesurfer@nmr.mgh.harvard.edumailto:freesurfer@nmr.mgh.harvard.edu freesurfer@nmr.mgh.harvard.edumailto:freesurfer@nmr.mgh.harvard.edu Subject: Re: [Freesurfer] highres fMRI selxavg error
Why are you combining them into one bold folder?
On 12/30/2019 9:09 AM, Nasiriavanaki, Zahra wrote: Dear Freesurfer experts
Happy Holidays! I have a subject with two highres fMRI scan sessions. Attached you can see an image, showing how I organized the sub-folders. I preprocessed the bold1 and bold2 and then copied the preprocessed files to "bold from both sessions" folder. I ran selxavg for bold1 and bold2 seperately and it went well. However, when I ran selxavg for "bold from both sessions" folder, it gives the below error. Could you please let me know what the problem is?Also if you have a better suggestion for how I should organize my subfolders/analysis, I appreciate that.
Thanks Mona
>> /autofs/cluster/freesurfer/centos7_x86_64/stable6/fsfast/toolbox/fast_selxavg3.m
/autofs/cluster/freesurfer/centos7_x86_64/stable6/fsfast/toolbox/fast_ldanaflac.m /autofs/cluster/freesurfer/centos7_x86_64/stable6/matlab/MRIread.m
starting fast_selxavg3b
#@# arap_7T_all ############################### /autofs/space/oprah_001/users/zn025/looming_7T/all_subjects/arap_7T_all ------------------------- $Id: fast_selxavg3b.m,v 1.4 2016/05/04 22:16:47 greve Exp $ ------------------------- outtop = /autofs/space/oprah_001/users/zn025/looming_7T/all_subjects Extension format = nii.gz INFO: key nSliceGroups unrecognized, line 12, skipping 1 app.mat 2 aw.mat 3 caw.mat 4 clo.mat 5 faw.mat 6 fem.mat 7 fm.mat 8 fmaw.mat 9 mal.mat 10 maw.mat 11 oaw.mat 12 oc.mat 13 ocaw.mat 14 ope.mat 15 wit.mat Excluding 6 points nruns = 15 autostimdur =
outanadir = /autofs/space/oprah_001/users/zn025/looming_7T/all_subjects/arap_7T_all/loom/loom_native.lh Excluding 6 points Excluding 0 points Excluding 1 points Excluding 5 points Excluding 8 points Excluding 3 points Excluding 8 points Excluding 4 points Excluding 6 points Excluding 10 points Excluding 5 points Excluding 5 points Excluding 5 points Excluding 5 points Excluding 5 points parfiles condition id list: 1 2 3 4 5 6 7 8 Found 41883/148565 (28.2) voxels in mask 1 Creating Design Matrix ... creation time = 0.040 sec DoMCFit = 1 ntptot = 1350, nX = 219, DOF = 1131 Saving X matrix to /autofs/space/oprah_001/users/zn025/looming_7T/all_subjects/arap_7T_all/loom/loom_native.lh/Xtmp.mat XCond = 1873.07 (normalized) Computing compensation for resdual AR1 bias 1 -0.5 -0.456169 (t=1.17104) 2 -0.25 -0.261165 (t=2.14097) 3 0 -0.076309 (t=2.3701) 4 0.25 0.0914527 (t=3.32529) 5 0.5 0.225506 (t=4.09175) AR1 Correction M: 0.138222 1.44983 Computing contrast matrices Computing contrasts Loading previous GLM fit Starting contrasts app J=1 ------------- Error using * Incorrect dimensions for matrix multiplication. Check that the number of columns in the first matrix matches the number of rows in the second matrix. To perform element wise multiplication, use '.*'.
Error in fast_fratiow (line 81) ces = C*beta;
Error in fast_selxavg3b (line 1153) fast_fratiow(betamat,X,rvarmat,C,acfsegmn,acfseg.vol(:));
ERROR: fast_selxavg3() failed\n
Zahra (Mona) Nasiriavanaki
Postdoctoral Research Fellow
Martinos Center for Biomedical Imaging
Massachusetts General Hospital
149 13th Street, 149-2615
Charlestown, MA, USA, 02129
_______________________________________________ Freesurfer mailing list Freesurfer@nmr.mgh.harvard.edumailto:Freesurfer@nmr.mgh.harvard.edu https://mail.nmr.mgh.harvard.edu/mailman/listinfo/freesurfer
It appears to be dying on this command mris_fwhm --mask /autofs/space/oprah_001/users/zn025/fearcond_7T/all_subjects/ylug_ass_sess01/bold/cond.lh/mask.nii Can you run it from the shell and send the terminal output?
On 1/2/2020 2:13 PM, Nasiriavanaki, Zahra wrote: Thanks a lot for your reply.
I have one more question about selxavg error and I appreciate if you could reply. I have another subject which has been scanned in 7T. When I run selxavg, it doesn't go through for the left and right hemi, but it gets perfectly done for subcortex. I attached the log file. We have collected the functional data in a slab of the brain containing subcortical areas and the insular cortex (at least this is what we aimed for). Looking at the data with tkregister2 command, I can't say if the insular cortex is included or not. Also, if it is included, then should we see the insular activation in cortical maps or subcortical ones? tkregister2 --s ylug_ass_sess01 --mov $all_subjects/ylug_ass_sess01/bold/010/template.nii.gz --surf --reg new.dat --regheader
Thanks a lot Mona
Zahra (Mona) Nasiriavanaki
Postdoctoral Research Fellow
Martinos Center for Biomedical Imaging
Massachusetts General Hospital
149 13th Street, 149-2615
Charlestown, MA, USA, 02129
________________________________ From: Greve, Douglas N.,Ph.D. DGREVE@mgh.harvard.edumailto:DGREVE@mgh.harvard.edu Sent: Thursday, January 2, 2020 1:18 PM To: Nasiriavanaki, Zahra ZNASIRIAVANAKI@mgh.harvard.edumailto:ZNASIRIAVANAKI@mgh.harvard.edu; freesurfer@nmr.mgh.harvard.edumailto:freesurfer@nmr.mgh.harvard.edu freesurfer@nmr.mgh.harvard.edumailto:freesurfer@nmr.mgh.harvard.edu Subject: Re: [Freesurfer] highres fMRI selxavg error
The easiest thing to do is to probably do the concatenation by hand, eg, mri_concat subj/sess01/analysis/contrast/ces.nii.gz subj/sess01/analysis/contrast/ces.nii.gz --o ces.sess01+sess02.nii.gz mri_concat subj/sess01/analysis/contrast/cesvar.nii.gz subj/sess01/analysis/contrast/cesvar.nii.gz --o cesvar.sess01+sess02.nii.gz
Create a dof file by adding together the dofs for each individual analysis as indicated by these values. Just put this number into a text file, eg, dof.sess01+sess02.dat cat subj/sess01/analysis/dof.dat subj/sess01/analysis/dof.dat
Create a mask fscalc subj/sess01/analysis/mask.nii.gz and subj/sess01/analysis/mask.nii.gz -o mask.sess01+sess02.nii.gz
Then run glmfit like mri_glmfit --surface subject lh --y ces.sess01+sess02.nii.gz --yffxvar cesvar.sess01+sess02.nii.gz --ffxdofdat dof.sess01+sess02.dat --mask mask.sess01+sess02.nii.gz --osgm --o ffx/subject/analysis/contrast/osgm
On 1/2/2020 11:20 AM, Nasiriavanaki, Zahra wrote: I want to concatenate the two sessions which I guess is the same as combining them statistically. Please note that it's all done in "self" space not fsaverage.
Zahra (Mona) Nasiriavanaki
Postdoctoral Research Fellow
Martinos Center for Biomedical Imaging
Massachusetts General Hospital
149 13th Street, 149-2615
Charlestown, MA, USA, 02129
________________________________ From: Greve, Douglas N.,Ph.D. DGREVE@mgh.harvard.edumailto:DGREVE@mgh.harvard.edu Sent: Thursday, January 2, 2020 11:46 AM To: Nasiriavanaki, Zahra ZNASIRIAVANAKI@mgh.harvard.edumailto:ZNASIRIAVANAKI@mgh.harvard.edu; freesurfer@nmr.mgh.harvard.edumailto:freesurfer@nmr.mgh.harvard.edu freesurfer@nmr.mgh.harvard.edumailto:freesurfer@nmr.mgh.harvard.edu Subject: Re: [Freesurfer] highres fMRI selxavg error
When you say you need to see them together, what does that mean? Are you computing contrasts between the two sessions or you just want to combine them statistically?
On 1/2/2020 10:07 AM, Nasiriavanaki, Zahra wrote: I need to concatenate the two sessions. We scanned the subjects twice with the same task. I need to see the maps for each session separately and also together.
Thanks
Zahra (Mona) Nasiriavanaki
Postdoctoral Research Fellow
Martinos Center for Biomedical Imaging
Massachusetts General Hospital
149 13th Street, 149-2615
Charlestown, MA, USA, 02129
________________________________ From: freesurfer-bounces@nmr.mgh.harvard.edumailto:freesurfer-bounces@nmr.mgh.harvard.edu freesurfer-bounces@nmr.mgh.harvard.edumailto:freesurfer-bounces@nmr.mgh.harvard.edu on behalf of Greve, Douglas N.,Ph.D. DGREVE@mgh.harvard.edumailto:DGREVE@mgh.harvard.edu Sent: Thursday, January 2, 2020 10:57 AM To: freesurfer@nmr.mgh.harvard.edumailto:freesurfer@nmr.mgh.harvard.edu freesurfer@nmr.mgh.harvard.edumailto:freesurfer@nmr.mgh.harvard.edu Subject: Re: [Freesurfer] highres fMRI selxavg error
Why are you combining them into one bold folder?
On 12/30/2019 9:09 AM, Nasiriavanaki, Zahra wrote: Dear Freesurfer experts
Happy Holidays! I have a subject with two highres fMRI scan sessions. Attached you can see an image, showing how I organized the sub-folders. I preprocessed the bold1 and bold2 and then copied the preprocessed files to "bold from both sessions" folder. I ran selxavg for bold1 and bold2 seperately and it went well. However, when I ran selxavg for "bold from both sessions" folder, it gives the below error. Could you please let me know what the problem is?Also if you have a better suggestion for how I should organize my subfolders/analysis, I appreciate that.
Thanks Mona
>> /autofs/cluster/freesurfer/centos7_x86_64/stable6/fsfast/toolbox/fast_selxavg3.m
/autofs/cluster/freesurfer/centos7_x86_64/stable6/fsfast/toolbox/fast_ldanaflac.m /autofs/cluster/freesurfer/centos7_x86_64/stable6/matlab/MRIread.m
starting fast_selxavg3b
#@# arap_7T_all ############################### /autofs/space/oprah_001/users/zn025/looming_7T/all_subjects/arap_7T_all ------------------------- $Id: fast_selxavg3b.m,v 1.4 2016/05/04 22:16:47 greve Exp $ ------------------------- outtop = /autofs/space/oprah_001/users/zn025/looming_7T/all_subjects Extension format = nii.gz INFO: key nSliceGroups unrecognized, line 12, skipping 1 app.mat 2 aw.mat 3 caw.mat 4 clo.mat 5 faw.mat 6 fem.mat 7 fm.mat 8 fmaw.mat 9 mal.mat 10 maw.mat 11 oaw.mat 12 oc.mat 13 ocaw.mat 14 ope.mat 15 wit.mat Excluding 6 points nruns = 15 autostimdur =
outanadir = /autofs/space/oprah_001/users/zn025/looming_7T/all_subjects/arap_7T_all/loom/loom_native.lh Excluding 6 points Excluding 0 points Excluding 1 points Excluding 5 points Excluding 8 points Excluding 3 points Excluding 8 points Excluding 4 points Excluding 6 points Excluding 10 points Excluding 5 points Excluding 5 points Excluding 5 points Excluding 5 points Excluding 5 points parfiles condition id list: 1 2 3 4 5 6 7 8 Found 41883/148565 (28.2) voxels in mask 1 Creating Design Matrix ... creation time = 0.040 sec DoMCFit = 1 ntptot = 1350, nX = 219, DOF = 1131 Saving X matrix to /autofs/space/oprah_001/users/zn025/looming_7T/all_subjects/arap_7T_all/loom/loom_native.lh/Xtmp.mat XCond = 1873.07 (normalized) Computing compensation for resdual AR1 bias 1 -0.5 -0.456169 (t=1.17104) 2 -0.25 -0.261165 (t=2.14097) 3 0 -0.076309 (t=2.3701) 4 0.25 0.0914527 (t=3.32529) 5 0.5 0.225506 (t=4.09175) AR1 Correction M: 0.138222 1.44983 Computing contrast matrices Computing contrasts Loading previous GLM fit Starting contrasts app J=1 ------------- Error using * Incorrect dimensions for matrix multiplication. Check that the number of columns in the first matrix matches the number of rows in the second matrix. To perform element wise multiplication, use '.*'.
Error in fast_fratiow (line 81) ces = C*beta;
Error in fast_selxavg3b (line 1153) fast_fratiow(betamat,X,rvarmat,C,acfsegmn,acfseg.vol(:));
ERROR: fast_selxavg3() failed\n
Zahra (Mona) Nasiriavanaki
Postdoctoral Research Fellow
Martinos Center for Biomedical Imaging
Massachusetts General Hospital
149 13th Street, 149-2615
Charlestown, MA, USA, 02129
_______________________________________________ Freesurfer mailing list Freesurfer@nmr.mgh.harvard.edumailto:Freesurfer@nmr.mgh.harvard.edu https://mail.nmr.mgh.harvard.edu/mailman/listinfo/freesurfer
I did run it from the shell and figured out what the problem was!
Thanks a lot!! Mona
Zahra (Mona) Nasiriavanaki
Postdoctoral Research Fellow
Martinos Center for Biomedical Imaging
Massachusetts General Hospital
149 13th Street, 149-2615
Charlestown, MA, USA, 02129
________________________________ From: Greve, Douglas N.,Ph.D. DGREVE@mgh.harvard.edu Sent: Thursday, January 2, 2020 3:25 PM To: Nasiriavanaki, Zahra ZNASIRIAVANAKI@mgh.harvard.edu; freesurfer@nmr.mgh.harvard.edu freesurfer@nmr.mgh.harvard.edu Subject: Re: [Freesurfer] highres fMRI selxavg error
It appears to be dying on this command mris_fwhm --mask /autofs/space/oprah_001/users/zn025/fearcond_7T/all_subjects/ylug_ass_sess01/bold/cond.lh/mask.nii Can you run it from the shell and send the terminal output?
On 1/2/2020 2:13 PM, Nasiriavanaki, Zahra wrote: Thanks a lot for your reply.
I have one more question about selxavg error and I appreciate if you could reply. I have another subject which has been scanned in 7T. When I run selxavg, it doesn't go through for the left and right hemi, but it gets perfectly done for subcortex. I attached the log file. We have collected the functional data in a slab of the brain containing subcortical areas and the insular cortex (at least this is what we aimed for). Looking at the data with tkregister2 command, I can't say if the insular cortex is included or not. Also, if it is included, then should we see the insular activation in cortical maps or subcortical ones? tkregister2 --s ylug_ass_sess01 --mov $all_subjects/ylug_ass_sess01/bold/010/template.nii.gz --surf --reg new.dat --regheader
Thanks a lot Mona
Zahra (Mona) Nasiriavanaki
Postdoctoral Research Fellow
Martinos Center for Biomedical Imaging
Massachusetts General Hospital
149 13th Street, 149-2615
Charlestown, MA, USA, 02129
________________________________ From: Greve, Douglas N.,Ph.D. DGREVE@mgh.harvard.edumailto:DGREVE@mgh.harvard.edu Sent: Thursday, January 2, 2020 1:18 PM To: Nasiriavanaki, Zahra ZNASIRIAVANAKI@mgh.harvard.edumailto:ZNASIRIAVANAKI@mgh.harvard.edu; freesurfer@nmr.mgh.harvard.edumailto:freesurfer@nmr.mgh.harvard.edu freesurfer@nmr.mgh.harvard.edumailto:freesurfer@nmr.mgh.harvard.edu Subject: Re: [Freesurfer] highres fMRI selxavg error
The easiest thing to do is to probably do the concatenation by hand, eg, mri_concat subj/sess01/analysis/contrast/ces.nii.gz subj/sess01/analysis/contrast/ces.nii.gz --o ces.sess01+sess02.nii.gz mri_concat subj/sess01/analysis/contrast/cesvar.nii.gz subj/sess01/analysis/contrast/cesvar.nii.gz --o cesvar.sess01+sess02.nii.gz
Create a dof file by adding together the dofs for each individual analysis as indicated by these values. Just put this number into a text file, eg, dof.sess01+sess02.dat cat subj/sess01/analysis/dof.dat subj/sess01/analysis/dof.dat
Create a mask fscalc subj/sess01/analysis/mask.nii.gz and subj/sess01/analysis/mask.nii.gz -o mask.sess01+sess02.nii.gz
Then run glmfit like mri_glmfit --surface subject lh --y ces.sess01+sess02.nii.gz --yffxvar cesvar.sess01+sess02.nii.gz --ffxdofdat dof.sess01+sess02.dat --mask mask.sess01+sess02.nii.gz --osgm --o ffx/subject/analysis/contrast/osgm
On 1/2/2020 11:20 AM, Nasiriavanaki, Zahra wrote: I want to concatenate the two sessions which I guess is the same as combining them statistically. Please note that it's all done in "self" space not fsaverage.
Zahra (Mona) Nasiriavanaki
Postdoctoral Research Fellow
Martinos Center for Biomedical Imaging
Massachusetts General Hospital
149 13th Street, 149-2615
Charlestown, MA, USA, 02129
________________________________ From: Greve, Douglas N.,Ph.D. DGREVE@mgh.harvard.edumailto:DGREVE@mgh.harvard.edu Sent: Thursday, January 2, 2020 11:46 AM To: Nasiriavanaki, Zahra ZNASIRIAVANAKI@mgh.harvard.edumailto:ZNASIRIAVANAKI@mgh.harvard.edu; freesurfer@nmr.mgh.harvard.edumailto:freesurfer@nmr.mgh.harvard.edu freesurfer@nmr.mgh.harvard.edumailto:freesurfer@nmr.mgh.harvard.edu Subject: Re: [Freesurfer] highres fMRI selxavg error
When you say you need to see them together, what does that mean? Are you computing contrasts between the two sessions or you just want to combine them statistically?
On 1/2/2020 10:07 AM, Nasiriavanaki, Zahra wrote: I need to concatenate the two sessions. We scanned the subjects twice with the same task. I need to see the maps for each session separately and also together.
Thanks
Zahra (Mona) Nasiriavanaki
Postdoctoral Research Fellow
Martinos Center for Biomedical Imaging
Massachusetts General Hospital
149 13th Street, 149-2615
Charlestown, MA, USA, 02129
________________________________ From: freesurfer-bounces@nmr.mgh.harvard.edumailto:freesurfer-bounces@nmr.mgh.harvard.edu freesurfer-bounces@nmr.mgh.harvard.edumailto:freesurfer-bounces@nmr.mgh.harvard.edu on behalf of Greve, Douglas N.,Ph.D. DGREVE@mgh.harvard.edumailto:DGREVE@mgh.harvard.edu Sent: Thursday, January 2, 2020 10:57 AM To: freesurfer@nmr.mgh.harvard.edumailto:freesurfer@nmr.mgh.harvard.edu freesurfer@nmr.mgh.harvard.edumailto:freesurfer@nmr.mgh.harvard.edu Subject: Re: [Freesurfer] highres fMRI selxavg error
Why are you combining them into one bold folder?
On 12/30/2019 9:09 AM, Nasiriavanaki, Zahra wrote: Dear Freesurfer experts
Happy Holidays! I have a subject with two highres fMRI scan sessions. Attached you can see an image, showing how I organized the sub-folders. I preprocessed the bold1 and bold2 and then copied the preprocessed files to "bold from both sessions" folder. I ran selxavg for bold1 and bold2 seperately and it went well. However, when I ran selxavg for "bold from both sessions" folder, it gives the below error. Could you please let me know what the problem is?Also if you have a better suggestion for how I should organize my subfolders/analysis, I appreciate that.
Thanks Mona
>> /autofs/cluster/freesurfer/centos7_x86_64/stable6/fsfast/toolbox/fast_selxavg3.m
/autofs/cluster/freesurfer/centos7_x86_64/stable6/fsfast/toolbox/fast_ldanaflac.m /autofs/cluster/freesurfer/centos7_x86_64/stable6/matlab/MRIread.m
starting fast_selxavg3b
#@# arap_7T_all ############################### /autofs/space/oprah_001/users/zn025/looming_7T/all_subjects/arap_7T_all ------------------------- $Id: fast_selxavg3b.m,v 1.4 2016/05/04 22:16:47 greve Exp $ ------------------------- outtop = /autofs/space/oprah_001/users/zn025/looming_7T/all_subjects Extension format = nii.gz INFO: key nSliceGroups unrecognized, line 12, skipping 1 app.mat 2 aw.mat 3 caw.mat 4 clo.mat 5 faw.mat 6 fem.mat 7 fm.mat 8 fmaw.mat 9 mal.mat 10 maw.mat 11 oaw.mat 12 oc.mat 13 ocaw.mat 14 ope.mat 15 wit.mat Excluding 6 points nruns = 15 autostimdur =
outanadir = /autofs/space/oprah_001/users/zn025/looming_7T/all_subjects/arap_7T_all/loom/loom_native.lh Excluding 6 points Excluding 0 points Excluding 1 points Excluding 5 points Excluding 8 points Excluding 3 points Excluding 8 points Excluding 4 points Excluding 6 points Excluding 10 points Excluding 5 points Excluding 5 points Excluding 5 points Excluding 5 points Excluding 5 points parfiles condition id list: 1 2 3 4 5 6 7 8 Found 41883/148565 (28.2) voxels in mask 1 Creating Design Matrix ... creation time = 0.040 sec DoMCFit = 1 ntptot = 1350, nX = 219, DOF = 1131 Saving X matrix to /autofs/space/oprah_001/users/zn025/looming_7T/all_subjects/arap_7T_all/loom/loom_native.lh/Xtmp.mat XCond = 1873.07 (normalized) Computing compensation for resdual AR1 bias 1 -0.5 -0.456169 (t=1.17104) 2 -0.25 -0.261165 (t=2.14097) 3 0 -0.076309 (t=2.3701) 4 0.25 0.0914527 (t=3.32529) 5 0.5 0.225506 (t=4.09175) AR1 Correction M: 0.138222 1.44983 Computing contrast matrices Computing contrasts Loading previous GLM fit Starting contrasts app J=1 ------------- Error using * Incorrect dimensions for matrix multiplication. Check that the number of columns in the first matrix matches the number of rows in the second matrix. To perform element wise multiplication, use '.*'.
Error in fast_fratiow (line 81) ces = C*beta;
Error in fast_selxavg3b (line 1153) fast_fratiow(betamat,X,rvarmat,C,acfsegmn,acfseg.vol(:));
ERROR: fast_selxavg3() failed\n
Zahra (Mona) Nasiriavanaki
Postdoctoral Research Fellow
Martinos Center for Biomedical Imaging
Massachusetts General Hospital
149 13th Street, 149-2615
Charlestown, MA, USA, 02129
_______________________________________________ Freesurfer mailing list Freesurfer@nmr.mgh.harvard.edumailto:Freesurfer@nmr.mgh.harvard.edu https://mail.nmr.mgh.harvard.edu/mailman/listinfo/freesurfer
Dear Freesurfer experts
Hi
I have a follow up question on this topic. I am getting this error "Error using /Matrix dimensions must agree. Error in fast_selxavg3b (line 566) RescaleFactor = flac0.inorm/gmean;", when I run selxavg for one of my subjects. I attached the log file and copied the terminal output below. Discussing it previously, we found out the problem being "global.meanval.dat and global.waveform.dat" files becoming empty after running selxavg! Listening to Doug's recommendation, I did run everything from scratch. After doing the preprocessing step, I realized the registration of functional on anatomical data is not correct using this command: "tkregister-sess -s $subj -fsd bold -per-run -bbr-sum". So I had to run "register-sess -s $subj -per-run -delete-dat -dof 6 -fsd bold -init-header -bbr-xopts bbr.xopts -d ." multiple times to make the registration correct (with --brute1max starting from 8 and every time I increased it. The last time I ran this command, --brute1max was set to 28). I got normal values for "tkregister-sess" and I ran selxavg and got the error that I attached. My question is: Do you think running "register-sess" multiple times is causing this issue? and I appreciate any suggestions about how to solve this issue.
P.S:This is highres functional MRI data; the functional data is only acquired from parietal and occipital lobes; out of several subjects; analysis is run in subject's native space.
Thanks a lot Mona
#@# osre_7T_all ############################### /autofs/space/oprah_001/users/zn025/looming_7T/all_subjects/osre_7T_all ------------------------- $Id: fast_selxavg3b.m,v 1.4 2016/05/04 22:16:47 greve Exp $ ------------------------- outtop = /autofs/space/oprah_001/users/zn025/looming_7T/all_subjects Extension format = nii.gz INFO: key nSliceGroups unrecognized, line 12, skipping 1 far.mat 2 far_vs_near.mat 3 far_vs_nodis.mat 4 near.mat 5 near_vs_far.mat 6 near_vs_nodis.mat 7 no_disparity.mat Excluding 5 points nruns = 8 autostimdur =
outanadir = /autofs/space/oprah_001/users/zn025/looming_7T/all_subjects/osre_7T_all/bold3/vis1_native.rh Excluding 5 points Excluding 6 points Excluding 6 points Excluding 7 points Excluding 8 points parfiles condition id list: 1 2 3 Found 1881/138983 ( 1.4) voxels in mask 1 Creating Design Matrix ... creation time = 0.020 sec DoMCFit = 1 ntptot = 640, nX = 107, DOF = 533 Saving X matrix to /autofs/space/oprah_001/users/zn025/looming_7T/all_subjects/osre_7T_all/bold3/vis1_native.rh/Xtmp.mat XCond = 448.137 (normalized) Computing compensation for resdual AR1 bias 1 -0.5 -0.4499 (t=0.059341) 2 -0.25 -0.255889 (t=0.147506) 3 0 -0.0718039 (t=0.175004) 4 0.25 0.0955918 (t=0.257562) 5 0.5 0.229844 (t=0.289084) AR1 Correction M: 0.131515 1.4543 Computing contrast matrices OLS Beta Pass run 1 t= 0.0 reading data ... 0.629912 Global Mean run 2 t= 0.8 reading data ... 0.666993 Global Mean run 3 t= 0.8 reading data ... 0.640162 Global Mean run 4 t= 0.8 reading data ... 0.646836 Global Mean run 5 t= 0.8 reading data ... 0.642453 Global Mean run 6 t= 0.8 reading data ... 0.64432 Global Mean run 7 t= 0.8 reading data ... 0.63922 Global Mean run 8 t= 0.8 reading data ... 0.63597 Global Mean Global In-Mask Mean = (35.6878) Error using / Matrix dimensions must agree.
Error in fast_selxavg3b (line 566) RescaleFactor = flac0.inorm/gmean;
ERROR: fast_selxavg3() failed\n
Zahra (Mona) Nasiriavanaki
Postdoctoral Research Fellow
Martinos Center for Biomedical Imaging
Massachusetts General Hospital
149 13th Street, 149-2615
Charlestown, MA, USA, 02129
________________________________ From: Nasiriavanaki, Zahra ZNASIRIAVANAKI@mgh.harvard.edu Sent: Thursday, January 2, 2020 3:40 PM To: Greve, Douglas N.,Ph.D. DGREVE@mgh.harvard.edu; freesurfer@nmr.mgh.harvard.edu freesurfer@nmr.mgh.harvard.edu Subject: Re: [Freesurfer] highres fMRI selxavg error
I did run it from the shell and figured out what the problem was!
Thanks a lot!! Mona
Zahra (Mona) Nasiriavanaki
Postdoctoral Research Fellow
Martinos Center for Biomedical Imaging
Massachusetts General Hospital
149 13th Street, 149-2615
Charlestown, MA, USA, 02129
________________________________ From: Greve, Douglas N.,Ph.D. DGREVE@mgh.harvard.edu Sent: Thursday, January 2, 2020 3:25 PM To: Nasiriavanaki, Zahra ZNASIRIAVANAKI@mgh.harvard.edu; freesurfer@nmr.mgh.harvard.edu freesurfer@nmr.mgh.harvard.edu Subject: Re: [Freesurfer] highres fMRI selxavg error
It appears to be dying on this command mris_fwhm --mask /autofs/space/oprah_001/users/zn025/fearcond_7T/all_subjects/ylug_ass_sess01/bold/cond.lh/mask.nii Can you run it from the shell and send the terminal output?
On 1/2/2020 2:13 PM, Nasiriavanaki, Zahra wrote: Thanks a lot for your reply.
I have one more question about selxavg error and I appreciate if you could reply. I have another subject which has been scanned in 7T. When I run selxavg, it doesn't go through for the left and right hemi, but it gets perfectly done for subcortex. I attached the log file. We have collected the functional data in a slab of the brain containing subcortical areas and the insular cortex (at least this is what we aimed for). Looking at the data with tkregister2 command, I can't say if the insular cortex is included or not. Also, if it is included, then should we see the insular activation in cortical maps or subcortical ones? tkregister2 --s ylug_ass_sess01 --mov $all_subjects/ylug_ass_sess01/bold/010/template.nii.gz --surf --reg new.dat --regheader
Thanks a lot Mona
Zahra (Mona) Nasiriavanaki
Postdoctoral Research Fellow
Martinos Center for Biomedical Imaging
Massachusetts General Hospital
149 13th Street, 149-2615
Charlestown, MA, USA, 02129
________________________________ From: Greve, Douglas N.,Ph.D. DGREVE@mgh.harvard.edumailto:DGREVE@mgh.harvard.edu Sent: Thursday, January 2, 2020 1:18 PM To: Nasiriavanaki, Zahra ZNASIRIAVANAKI@mgh.harvard.edumailto:ZNASIRIAVANAKI@mgh.harvard.edu; freesurfer@nmr.mgh.harvard.edumailto:freesurfer@nmr.mgh.harvard.edu freesurfer@nmr.mgh.harvard.edumailto:freesurfer@nmr.mgh.harvard.edu Subject: Re: [Freesurfer] highres fMRI selxavg error
The easiest thing to do is to probably do the concatenation by hand, eg, mri_concat subj/sess01/analysis/contrast/ces.nii.gz subj/sess01/analysis/contrast/ces.nii.gz --o ces.sess01+sess02.nii.gz mri_concat subj/sess01/analysis/contrast/cesvar.nii.gz subj/sess01/analysis/contrast/cesvar.nii.gz --o cesvar.sess01+sess02.nii.gz
Create a dof file by adding together the dofs for each individual analysis as indicated by these values. Just put this number into a text file, eg, dof.sess01+sess02.dat cat subj/sess01/analysis/dof.dat subj/sess01/analysis/dof.dat
Create a mask fscalc subj/sess01/analysis/mask.nii.gz and subj/sess01/analysis/mask.nii.gz -o mask.sess01+sess02.nii.gz
Then run glmfit like mri_glmfit --surface subject lh --y ces.sess01+sess02.nii.gz --yffxvar cesvar.sess01+sess02.nii.gz --ffxdofdat dof.sess01+sess02.dat --mask mask.sess01+sess02.nii.gz --osgm --o ffx/subject/analysis/contrast/osgm
On 1/2/2020 11:20 AM, Nasiriavanaki, Zahra wrote: I want to concatenate the two sessions which I guess is the same as combining them statistically. Please note that it's all done in "self" space not fsaverage.
Zahra (Mona) Nasiriavanaki
Postdoctoral Research Fellow
Martinos Center for Biomedical Imaging
Massachusetts General Hospital
149 13th Street, 149-2615
Charlestown, MA, USA, 02129
________________________________ From: Greve, Douglas N.,Ph.D. DGREVE@mgh.harvard.edumailto:DGREVE@mgh.harvard.edu Sent: Thursday, January 2, 2020 11:46 AM To: Nasiriavanaki, Zahra ZNASIRIAVANAKI@mgh.harvard.edumailto:ZNASIRIAVANAKI@mgh.harvard.edu; freesurfer@nmr.mgh.harvard.edumailto:freesurfer@nmr.mgh.harvard.edu freesurfer@nmr.mgh.harvard.edumailto:freesurfer@nmr.mgh.harvard.edu Subject: Re: [Freesurfer] highres fMRI selxavg error
When you say you need to see them together, what does that mean? Are you computing contrasts between the two sessions or you just want to combine them statistically?
On 1/2/2020 10:07 AM, Nasiriavanaki, Zahra wrote: I need to concatenate the two sessions. We scanned the subjects twice with the same task. I need to see the maps for each session separately and also together.
Thanks
Zahra (Mona) Nasiriavanaki
Postdoctoral Research Fellow
Martinos Center for Biomedical Imaging
Massachusetts General Hospital
149 13th Street, 149-2615
Charlestown, MA, USA, 02129
________________________________ From: freesurfer-bounces@nmr.mgh.harvard.edumailto:freesurfer-bounces@nmr.mgh.harvard.edu freesurfer-bounces@nmr.mgh.harvard.edumailto:freesurfer-bounces@nmr.mgh.harvard.edu on behalf of Greve, Douglas N.,Ph.D. DGREVE@mgh.harvard.edumailto:DGREVE@mgh.harvard.edu Sent: Thursday, January 2, 2020 10:57 AM To: freesurfer@nmr.mgh.harvard.edumailto:freesurfer@nmr.mgh.harvard.edu freesurfer@nmr.mgh.harvard.edumailto:freesurfer@nmr.mgh.harvard.edu Subject: Re: [Freesurfer] highres fMRI selxavg error
Why are you combining them into one bold folder?
On 12/30/2019 9:09 AM, Nasiriavanaki, Zahra wrote: Dear Freesurfer experts
Happy Holidays! I have a subject with two highres fMRI scan sessions. Attached you can see an image, showing how I organized the sub-folders. I preprocessed the bold1 and bold2 and then copied the preprocessed files to "bold from both sessions" folder. I ran selxavg for bold1 and bold2 seperately and it went well. However, when I ran selxavg for "bold from both sessions" folder, it gives the below error. Could you please let me know what the problem is?Also if you have a better suggestion for how I should organize my subfolders/analysis, I appreciate that.
Thanks Mona
>> /autofs/cluster/freesurfer/centos7_x86_64/stable6/fsfast/toolbox/fast_selxavg3.m
/autofs/cluster/freesurfer/centos7_x86_64/stable6/fsfast/toolbox/fast_ldanaflac.m /autofs/cluster/freesurfer/centos7_x86_64/stable6/matlab/MRIread.m
starting fast_selxavg3b
#@# arap_7T_all ############################### /autofs/space/oprah_001/users/zn025/looming_7T/all_subjects/arap_7T_all ------------------------- $Id: fast_selxavg3b.m,v 1.4 2016/05/04 22:16:47 greve Exp $ ------------------------- outtop = /autofs/space/oprah_001/users/zn025/looming_7T/all_subjects Extension format = nii.gz INFO: key nSliceGroups unrecognized, line 12, skipping 1 app.mat 2 aw.mat 3 caw.mat 4 clo.mat 5 faw.mat 6 fem.mat 7 fm.mat 8 fmaw.mat 9 mal.mat 10 maw.mat 11 oaw.mat 12 oc.mat 13 ocaw.mat 14 ope.mat 15 wit.mat Excluding 6 points nruns = 15 autostimdur =
outanadir = /autofs/space/oprah_001/users/zn025/looming_7T/all_subjects/arap_7T_all/loom/loom_native.lh Excluding 6 points Excluding 0 points Excluding 1 points Excluding 5 points Excluding 8 points Excluding 3 points Excluding 8 points Excluding 4 points Excluding 6 points Excluding 10 points Excluding 5 points Excluding 5 points Excluding 5 points Excluding 5 points Excluding 5 points parfiles condition id list: 1 2 3 4 5 6 7 8 Found 41883/148565 (28.2) voxels in mask 1 Creating Design Matrix ... creation time = 0.040 sec DoMCFit = 1 ntptot = 1350, nX = 219, DOF = 1131 Saving X matrix to /autofs/space/oprah_001/users/zn025/looming_7T/all_subjects/arap_7T_all/loom/loom_native.lh/Xtmp.mat XCond = 1873.07 (normalized) Computing compensation for resdual AR1 bias 1 -0.5 -0.456169 (t=1.17104) 2 -0.25 -0.261165 (t=2.14097) 3 0 -0.076309 (t=2.3701) 4 0.25 0.0914527 (t=3.32529) 5 0.5 0.225506 (t=4.09175) AR1 Correction M: 0.138222 1.44983 Computing contrast matrices Computing contrasts Loading previous GLM fit Starting contrasts app J=1 ------------- Error using * Incorrect dimensions for matrix multiplication. Check that the number of columns in the first matrix matches the number of rows in the second matrix. To perform element wise multiplication, use '.*'.
Error in fast_fratiow (line 81) ces = C*beta;
Error in fast_selxavg3b (line 1153) fast_fratiow(betamat,X,rvarmat,C,acfsegmn,acfseg.vol(:));
ERROR: fast_selxavg3() failed\n
Zahra (Mona) Nasiriavanaki
Postdoctoral Research Fellow
Martinos Center for Biomedical Imaging
Massachusetts General Hospital
149 13th Street, 149-2615
Charlestown, MA, USA, 02129
_______________________________________________ Freesurfer mailing list Freesurfer@nmr.mgh.harvard.edumailto:Freesurfer@nmr.mgh.harvard.edu https://mail.nmr.mgh.harvard.edu/mailman/listinfo/freesurfer
freesurfer@nmr.mgh.harvard.edu
-
 Greve, Douglas N.,Ph.D.
Greve, Douglas N.,Ph.D. -
 Nasiriavanaki, Zahra
Nasiriavanaki, Zahra