
Dear freesurfer experts,
I experienced some problems with the segmentation of my T1 scans, as both the white surface and the pial surface are placed incorrectly (see attached images). This seems to me to be a too big difference with what it should look like, that I think could not be fixed with the standard troubleshooting strategies.
Do you have any solutions for this? Is it for instance possible to make changes to the recon-all script to shift the surfaces?
Thanks in advance,
Doety
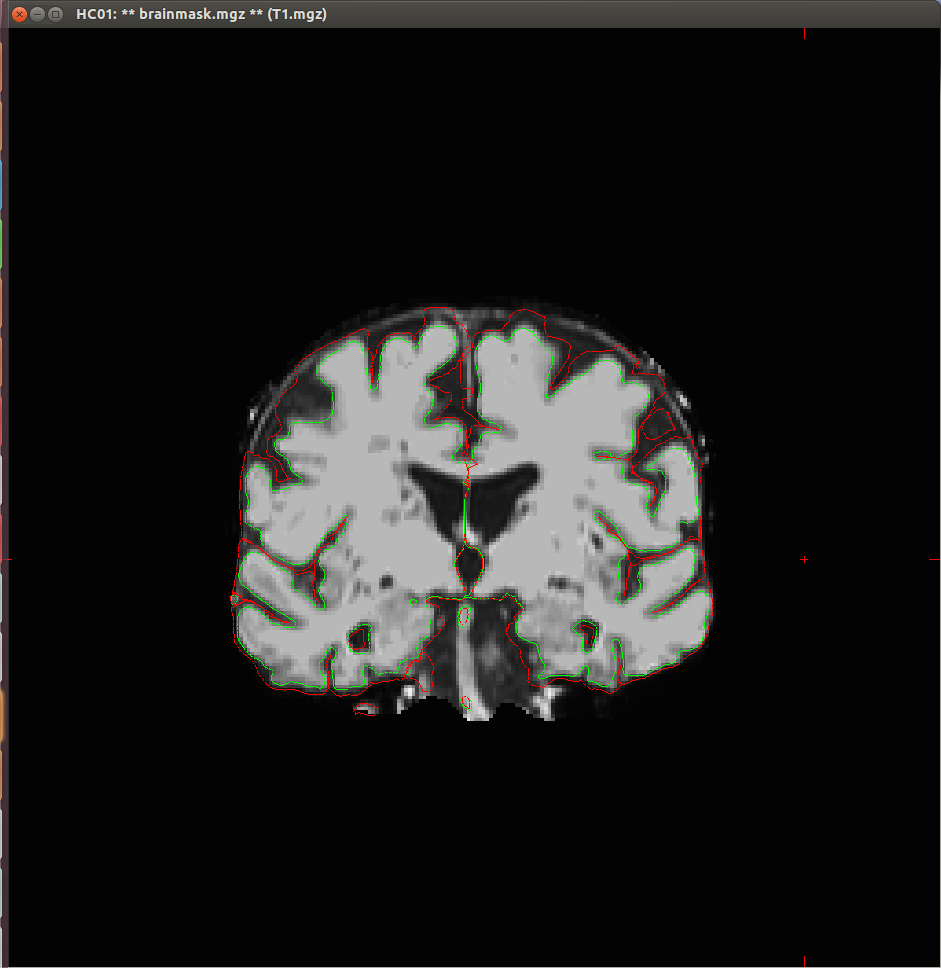{kind=link}
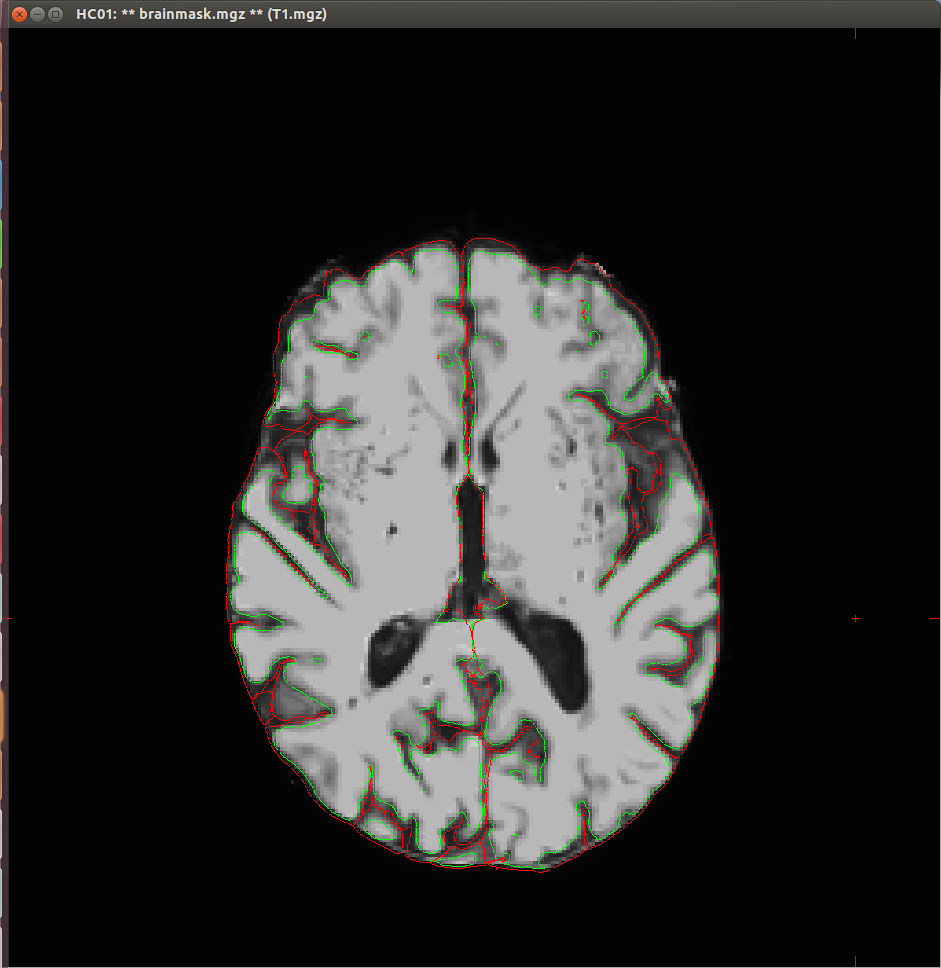{kind=link}

Hi Doety
yes, that looks pretty bad. Can you tell us about the acquisition? What resolution/field strength/scan type was it?
If you upload the subject we will take a look
cheers Bruce On Wed, 9 Jul 2014, Doety Prins wrote:
Dear freesurfer experts,
I experienced some problems with the segmentation of my T1 scans, as both the white surface and the pial surface are placed incorrectly (see attached images). This seems to me to be a too big difference with what it should look like, that I think could not be fixed with the standard troubleshooting strategies.
Do you have any solutions for this? Is it for instance possible to make changes to the recon-all script to shift the surfaces?
Thanks in advance,
Doety
Hi Bruce,
Thanks for your reply. In my previous e-mail I already tried to attach one of my subjects, but got the response that the message was too big, and therefore it was rejected. So how should I upload the image? The images are T1-weighted, 3D, acquired with a 3T scanner, resolution 256 256.
I discovered that FSL does a proper segmentation on this subjects, so I was looking for a way to import these segments from FSL into the Freesurfer pipeline, but I didn't succeed so far. I used the white matter segment from FSL as wm.mgz (in a subject in which I already ran recon-all), I used mri_vol2vol to get this segment in the right space, and normalized with mri_normalize. Then I rerun recon-all with -autorecon2-wm. But it gives me the error message: 'ERROR: mri_segment-MRIcheckVolDims: volume1 depth=160 != volume2 depth=256.' So apparently, the wm.mgz still doesn't have the right size. Do you have any experience with this? Or any ideas about this?
Best regards,
Doety
On 9 jul. 2014, at 15:00, Bruce Fischl fischl@nmr.mgh.harvard.edu wrote:
Hi Doety
yes, that looks pretty bad. Can you tell us about the acquisition? What resolution/field strength/scan type was it?
If you upload the subject we will take a look
cheers Bruce On Wed, 9 Jul 2014, Doety Prins wrote:
Dear freesurfer experts,
I experienced some problems with the segmentation of my T1 scans, as both the white surface and the pial surface are placed incorrectly (see attached images). This seems to me to be a too big difference with what it should look like, that I think could not be fixed with the standard troubleshooting strategies.
Do you have any solutions for this? Is it for instance possible to make changes to the recon-all script to shift the surfaces?
Thanks in advance,
Doety
Freesurfer mailing list Freesurfer@nmr.mgh.harvard.edu https://mail.nmr.mgh.harvard.edu/mailman/listinfo/freesurfer
The information in this e-mail is intended only for the person to whom it is addressed. If you believe this e-mail was sent to you in error and the e-mail contains patient information, please contact the Partners Compliance HelpLine at http://www.partners.org/complianceline . If the e-mail was sent to you in error but does not contain patient information, please contact the sender and properly dispose of the e-mail.

Hi Doety, Consider these options for uploading data: https://surfer.nmr.mgh.harvard.edu/fswiki/FtpFileExchange https://www.nmr.mgh.harvard.edu/facility/filedrop/index.html
I think recon-all is expecting your wm.mgz to be conformed or 256^3. Something like: mri_convert wm.mgz wm.conform.mgz --conform
You would need to rename wm.conform.mgz to wm.mgz, after renaming or saving a copy of the current non-conformed wm mask you got from FSL. -Louis
On Thu, 10 Jul 2014, Doety Prins wrote:
Hi Bruce,
Thanks for your reply. In my previous e-mail I already tried to attach one of my subjects, but got the response that the message was too big, and therefore it was rejected. So how should I upload the image? The images are T1-weighted, 3D, acquired with a 3T scanner, resolution 256 256.
I discovered that FSL does a proper segmentation on this subjects, so I was looking for a way to import these segments from FSL into the Freesurfer pipeline, but I didn't succeed so far. I used the white matter segment from FSL as wm.mgz (in a subject in which I already ran recon-all), I used mri_vol2vol to get this segment in the right space, and normalized with mri_normalize. Then I rerun recon-all with -autorecon2-wm. But it gives me the error message: 'ERROR: mri_segment-MRIcheckVolDims: volume1 depth=160 != volume2 depth=256.' So apparently, the wm.mgz still doesn't have the right size. Do you have any experience with this? Or any ideas about this?
Best regards,
Doety
On 9 jul. 2014, at 15:00, Bruce Fischl fischl@nmr.mgh.harvard.edu wrote:
Hi Doety
yes, that looks pretty bad. Can you tell us about the acquisition? What resolution/field strength/scan type was it?
If you upload the subject we will take a look
cheers Bruce On Wed, 9 Jul 2014, Doety Prins wrote:
Dear freesurfer experts,
I experienced some problems with the segmentation of my T1 scans, as both the white surface and the pial surface are placed incorrectly (see attached images). This seems to me to be a too big difference with what it should look like, that I think could not be fixed with the standard troubleshooting strategies.
Do you have any solutions for this? Is it for instance possible to make changes to the recon-all script to shift the surfaces?
Thanks in advance,
Doety
Freesurfer mailing list Freesurfer@nmr.mgh.harvard.edu https://mail.nmr.mgh.harvard.edu/mailman/listinfo/freesurfer
The information in this e-mail is intended only for the person to whom it is addressed. If you believe this e-mail was sent to you in error and the e-mail contains patient information, please contact the Partners Compliance HelpLine at http://www.partners.org/complianceline . If the e-mail was sent to you in error but does not contain patient information, please contact the sender and properly dispose of the e-mail.
Freesurfer mailing list Freesurfer@nmr.mgh.harvard.edu https://mail.nmr.mgh.harvard.edu/mailman/listinfo/freesurfer
Hi Doety
that is a very low contrast-to-noise image. Can you give us more details on the acquisition? The matrix was 256x256, what is the slice thickness? And the other parameter (sequence name? TR/TR/flip angle? acceleration?)
cheers Bruce
On Thu, 10 Jul 2014, Doety Prins wrote:
Hi Bruce,
Thanks for your reply. In my previous e-mail I already tried to attach
one of my subjects, but got the response that the message was too big, and therefore it was rejected. So how should I upload the image? The images are T1-weighted, 3D, acquired with a 3T scanner, resolution 256 256.
I discovered that FSL does a proper segmentation on this subjects, so I was looking for a way to import these segments from FSL into the Freesurfer pipeline, but I didn't succeed so far. I used the white matter segment from FSL as wm.mgz (in a subject in which I already ran recon-all), I used mri_vol2vol to get this segment in the right space, and normalized with mri_normalize. Then I rerun recon-all with -autorecon2-wm. But it gives me the error message: 'ERROR: mri_segment-MRIcheckVolDims: volume1 depth=160 != volume2 depth=256.' So apparently, the wm.mgz still doesn't have the right size. Do you have any experience with this? Or any ideas about this?
Best regards,
Doety
On 9 jul. 2014, at 15:00, Bruce Fischl fischl@nmr.mgh.harvard.edu wrote:
Hi Doety
yes, that looks pretty bad. Can you tell us about the acquisition? What resolution/field strength/scan type was it?
If you upload the subject we will take a look
cheers Bruce On Wed, 9 Jul 2014, Doety Prins wrote:
Dear freesurfer experts,
I experienced some problems with the segmentation of my T1 scans, as both the white surface and the pial surface are placed incorrectly (see attached images). This seems to me to be a too big difference with what it should look like, that I think could not be fixed with the standard troubleshooting strategies.
Do you have any solutions for this? Is it for instance possible to make changes to the recon-all script to shift the surfaces?
Thanks in advance,
Doety
Freesurfer mailing list Freesurfer@nmr.mgh.harvard.edu https://mail.nmr.mgh.harvard.edu/mailman/listinfo/freesurfer
The information in this e-mail is intended only for the person to whom it is addressed. If you believe this e-mail was sent to you in error and the e-mail contains patient information, please contact the Partners Compliance HelpLine at http://www.partners.org/complianceline . If the e-mail was sent to you in error but does not contain patient information, please contact the sender and properly dispose of the e-mail.
Freesurfer mailing list Freesurfer@nmr.mgh.harvard.edu https://mail.nmr.mgh.harvard.edu/mailman/listinfo/freesurfer
slice thickness: 2 mm Repetition time: 25 ms (I think this might have caused the low contrast) flip angle: 30 degrees I'm sorry, can't find any data on acceleration
Doety
On 11 jul. 2014, at 15:10, Bruce Fischl fischl@nmr.mgh.harvard.edu wrote:
Hi Doety
that is a very low contrast-to-noise image. Can you give us more details on the acquisition? The matrix was 256x256, what is the slice thickness? And the other parameter (sequence name? TR/TR/flip angle? acceleration?)
cheers Bruce
On Thu, 10 Jul 2014, Doety Prins wrote:
Hi Bruce,
Thanks for your reply. In my previous e-mail I already tried to attach
one of my subjects, but got the response that the message was too big, and therefore it was rejected. So how should I upload the image? The images are T1-weighted, 3D, acquired with a 3T scanner, resolution 256 256.
I discovered that FSL does a proper segmentation on this subjects, so I was looking for a way to import these segments from FSL into the Freesurfer pipeline, but I didn't succeed so far. I used the white matter segment from FSL as wm.mgz (in a subject in which I already ran recon-all), I used mri_vol2vol to get this segment in the right space, and normalized with mri_normalize. Then I rerun recon-all with -autorecon2-wm. But it gives me the error message: 'ERROR: mri_segment-MRIcheckVolDims: volume1 depth=160 != volume2 depth=256.' So apparently, the wm.mgz still doesn't have the right size. Do you have any experience with this? Or any ideas about this?
Best regards,
Doety
On 9 jul. 2014, at 15:00, Bruce Fischl fischl@nmr.mgh.harvard.edu wrote:
Hi Doety
yes, that looks pretty bad. Can you tell us about the acquisition? What resolution/field strength/scan type was it?
If you upload the subject we will take a look
cheers Bruce On Wed, 9 Jul 2014, Doety Prins wrote:
Dear freesurfer experts,
I experienced some problems with the segmentation of my T1 scans, as both the white surface and the pial surface are placed incorrectly (see attached images). This seems to me to be a too big difference with what it should look like, that I think could not be fixed with the standard troubleshooting strategies.
Do you have any solutions for this? Is it for instance possible to make changes to the recon-all script to shift the surfaces?
Thanks in advance,
Doety
Freesurfer mailing list Freesurfer@nmr.mgh.harvard.edu https://mail.nmr.mgh.harvard.edu/mailman/listinfo/freesurfer
The information in this e-mail is intended only for the person to whom it is addressed. If you believe this e-mail was sent to you in error and the e-mail contains patient information, please contact the Partners Compliance HelpLine at http://www.partners.org/complianceline . If the e-mail was sent to you in error but does not contain patient information, please contact the sender and properly dispose of the e-mail.
Freesurfer mailing list Freesurfer@nmr.mgh.harvard.edu https://mail.nmr.mgh.harvard.edu/mailman/listinfo/freesurfer
Freesurfer mailing list Freesurfer@nmr.mgh.harvard.edu https://mail.nmr.mgh.harvard.edu/mailman/listinfo/freesurfer
the 2mm slice thickness is going to be problematic and is probably why you lose so much cortical constrast. Typically we don't recommend using anything more than 1.5 and really no reason these days not to get closer to 1mm (for example, you can get a very nice 1.25mmx1.25x1mm mprage in a bit over 2 minutes)
cheers Bruce
On Fri, 11 Jul 2014, Doety Prins wrote:
slice thickness: 2 mm Repetition time: 25 ms (I think this might have caused the low contrast) flip angle: 30 degrees I'm sorry, can't find any data on acceleration
Doety
On 11 jul. 2014, at 15:10, Bruce Fischl fischl@nmr.mgh.harvard.edu wrote:
Hi Doety
that is a very low contrast-to-noise image. Can you give us more details on the acquisition? The matrix was 256x256, what is the slice thickness? And the other parameter (sequence name? TR/TR/flip angle? acceleration?)
cheers Bruce
On Thu, 10 Jul 2014, Doety Prins wrote:
Hi Bruce,
Thanks for your reply. In my previous e-mail I already tried to attach
one of my subjects, but got the response that the message was too big, and therefore it was rejected. So how should I upload the image? The images are T1-weighted, 3D, acquired with a 3T scanner, resolution 256 256.
I discovered that FSL does a proper segmentation on this subjects, so I was looking for a way to import these segments from FSL into the Freesurfer pipeline, but I didn't succeed so far. I used the white matter segment from FSL as wm.mgz (in a subject in which I already ran recon-all), I used mri_vol2vol to get this segment in the right space, and normalized with mri_normalize. Then I rerun recon-all with -autorecon2-wm. But it gives me the error message: 'ERROR: mri_segment-MRIcheckVolDims: volume1 depth=160 != volume2 depth=256.' So apparently, the wm.mgz still doesn't have the right size. Do you have any experience with this? Or any ideas about this?
Best regards,
Doety
On 9 jul. 2014, at 15:00, Bruce Fischl fischl@nmr.mgh.harvard.edu wrote:
Hi Doety
yes, that looks pretty bad. Can you tell us about the acquisition? What resolution/field strength/scan type was it?
If you upload the subject we will take a look
cheers Bruce On Wed, 9 Jul 2014, Doety Prins wrote:
Dear freesurfer experts,
I experienced some problems with the segmentation of my T1 scans, as both the white surface and the pial surface are placed incorrectly (see attached images). This seems to me to be a too big difference with what it should look like, that I think could not be fixed with the standard troubleshooting strategies.
Do you have any solutions for this? Is it for instance possible to make changes to the recon-all script to shift the surfaces?
Thanks in advance,
Doety
Freesurfer mailing list Freesurfer@nmr.mgh.harvard.edu https://mail.nmr.mgh.harvard.edu/mailman/listinfo/freesurfer
The information in this e-mail is intended only for the person to whom it is addressed. If you believe this e-mail was sent to you in error and the e-mail contains patient information, please contact the Partners Compliance HelpLine at http://www.partners.org/complianceline . If the e-mail was sent to you in error but does not contain patient information, please contact the sender and properly dispose of the e-mail.
Freesurfer mailing list Freesurfer@nmr.mgh.harvard.edu https://mail.nmr.mgh.harvard.edu/mailman/listinfo/freesurfer
Freesurfer mailing list Freesurfer@nmr.mgh.harvard.edu https://mail.nmr.mgh.harvard.edu/mailman/listinfo/freesurfer
Freesurfer mailing list Freesurfer@nmr.mgh.harvard.edu https://mail.nmr.mgh.harvard.edu/mailman/listinfo/freesurfer
Hi Bruce,
Thanks for your reply. Yes I realize now that this is causing me the troubles. But since fsl is able to do a good segmentation, I think Freesurfer should also be able to do this?
I am now using MRI_segment, in which I want to set new limits for the white matter, but it doesn't seem to work yet. I use wm.mgz as input volume and output volume something like wm.new.mgz, and -wm_low and -wm_hi with different values as optional arguments. But what file should be included as 'classifier file'? I couldn't find that anywhere in the description.
Thanks in advance for your help again!
Doety
Op vrijdag 11 juli 2014 heeft Bruce Fischl fischl@nmr.mgh.harvard.edu het volgende geschreven:
the 2mm slice thickness is going to be problematic and is probably why you lose so much cortical constrast. Typically we don't recommend using anything more than 1.5 and really no reason these days not to get closer
to
1mm (for example, you can get a very nice 1.25mmx1.25x1mm mprage in a bit over 2 minutes)
cheers Bruce
On Fri, 11 Jul 2014, Doety Prins wrote:
slice thickness: 2 mm Repetition time: 25 ms (I think this might have caused the low contrast) flip angle: 30 degrees I'm sorry, can't find any data on acceleration
Doety
On 11 jul. 2014, at 15:10, Bruce Fischl fischl@nmr.mgh.harvard.edu
wrote:
Hi Doety
that is a very low contrast-to-noise image. Can you give us more details on the acquisition? The matrix was 256x256, what is the slice thickness? And the other parameter (sequence name? TR/TR/flip angle? acceleration?)
cheers Bruce
On Thu, 10 Jul 2014, Doety Prins wrote:
Hi Bruce,
Thanks for your reply. In my previous e-mail I already tried to attach
one of my subjects, but got the response that the message was too big,
and
therefore it was rejected. So how should I upload the image? The images
are
T1-weighted, 3D, acquired with a 3T scanner, resolution 256 256.
I discovered that FSL does a proper segmentation on this subjects, so
I was looking for a way to import these segments from FSL into the Freesurfer pipeline, but I didn't succeed so far. I used the white matter segment from FSL as wm.mgz (in a subject in which I already ran recon-all), I used mri_vol2vol to get this segment in the right space, and normalized with mri_normalize. Then I rerun recon-all with -autorecon2-wm. But it gives me the error message: 'ERROR: mri_segment-MRIcheckVolDims: volume1 depth=160 != volume2 depth=256.' So apparently, the wm.mgz still doesn't have the right size. Do you have any experience with this? Or any ideas about this?
Best regards,
Doety
On 9 jul. 2014, at 15:00, Bruce Fischl fischl@nmr.mgh.harvard.edu
wrote:
Hi Doety
yes, that looks pretty bad. Can you tell us about the acquisition?
What
resolution/field strength/scan type was it?
If you upload the subject we will take a look
cheers Bruce On Wed, 9 Jul 2014, Doety Prins wrote:
> Dear freesurfer experts, > > I experienced some problems with the segmentation of my T1 scans,
as both the white surface and the pial surface are placed incorrectly (see attached images). This seems to me to be a too big difference with what it should look like, that I think could not be fixed with the standard troubleshooting strategies.
> > Do you have any solutions for this? Is it for instance possible to
make changes to the recon-all script to shift the surfaces?
> > Thanks in advance, > > Doety
Freesurfer mailing list Freesurfer@nmr.mgh.harvard.edu https://mail.nmr.mgh.harvard.edu/mailman/listinfo/freesurfer
The information in this e-mail is intended only for the person to
whom it is
addressed. If you believe this e-mail was sent to you in error and
the e-mail
contains patient information, please contact the Partners Compliance
HelpLine at
http://www.partners.org/complianceline . If the e-mail was sent to
you in error
but does not contain patient information, please contact the sender
and properly
dispose of the e-mail.
Freesurfer mailing list Freesurfer@nmr.mgh.harvard.edu https://mail.nmr.mgh.harvard.edu/mailman/listinfo/freesurfer
Freesurfer mailing list Freesurfer@nmr.mgh.harvard.edu https://mail.nmr.mgh.harvard.edu/mailman/listinfo/freesurfer
Freesurfer mailing list Freesurfer@nmr.mgh.harvard.edu https://mail.nmr.mgh.harvard.edu/mailman/listinfo/freesurfer
Freesurfer mailing list Freesurfer@nmr.mgh.harvard.edu https://mail.nmr.mgh.harvard.edu/mailman/listinfo/freesurfer
Hi Doety
our segmentation assumes we can resolve cortex as it used an adaptive filtering that estimates the orientation of the gray/white boundary in spatial neighborhoods. It may be more sensitive to low resolution than FSL. Not sure if anyone has a script for importing FSL segmentations into our processing pipeline, but someone might.....
cheers Bruce
On Tue, 15 Jul 2014, Doety Prins wrote:
Hi Bruce,
Thanks for your reply. Yes I realize now that this is causing me the troubles. But since fsl is able to do a good segmentation, I think Freesurfer should also be able to do this?
I am now using MRI_segment, in which I want to set new limits for the white matter, but it doesn't seem to work yet. I use wm.mgz as input volume and output volume something like wm.new.mgz, and -wm_low and -wm_hi with different values as optional arguments. But what file should be included as 'classifier file'? I couldn't find that anywhere in the description.
Thanks in advance for your help again!
Doety
Op vrijdag 11 juli 2014 heeft Bruce Fischl fischl@nmr.mgh.harvard.edu het volgende geschreven:
the 2mm slice thickness is going to be problematic and is probably why you lose so much cortical constrast. Typically we don't recommend using anything more than 1.5 and really no reason these days not to get closer
to
1mm (for example, you can get a very nice 1.25mmx1.25x1mm mprage in a bit over 2 minutes)
cheers Bruce
On Fri, 11 Jul 2014, Doety Prins wrote:
slice thickness: 2 mm Repetition time: 25 ms (I think this might have caused the low contrast) flip angle: 30 degrees I'm sorry, can't find any data on acceleration
Doety
On 11 jul. 2014, at 15:10, Bruce Fischl fischl@nmr.mgh.harvard.edu
wrote:
Hi Doety
that is a very low contrast-to-noise image. Can you give us more details on the acquisition? The matrix was 256x256, what is the slice thickness? And the other parameter (sequence name? TR/TR/flip angle? acceleration?)
cheers Bruce
On Thu, 10 Jul 2014, Doety Prins wrote:
Hi Bruce,
Thanks for your reply. In my previous e-mail I already tried to attach
one of my subjects, but got the response that the message was too big,
and
therefore it was rejected. So how should I upload the image? The images
are
T1-weighted, 3D, acquired with a 3T scanner, resolution 256 256.
I discovered that FSL does a proper segmentation on this subjects, so I
was looking for a way to import these segments from FSL into the Freesurfer pipeline, but I didn't succeed so far. I used the white matter segment from FSL as wm.mgz (in a subject in which I already ran recon-all), I used mri_vol2vol to get this segment in the right space, and normalized with mri_normalize. Then I rerun recon-all with -autorecon2-wm. But it gives me the error message: 'ERROR: mri_segment-MRIcheckVolDims: volume1 depth=160 != volume2 depth=256.' So apparently, the wm.mgz still doesn't have the right size. Do you have any experience with this? Or any ideas about this?
Best regards,
Doety
On 9 jul. 2014, at 15:00, Bruce Fischl fischl@nmr.mgh.harvard.edu
wrote:
Hi Doety
yes, that looks pretty bad. Can you tell us about the acquisition?
What
resolution/field strength/scan type was it?
If you upload the subject we will take a look
cheers Bruce On Wed, 9 Jul 2014, Doety Prins wrote:
>> Dear freesurfer experts, >> >> I experienced some problems with the segmentation of my T1 scans, as
both the white surface and the pial surface are placed incorrectly (see attached images). This seems to me to be a too big difference with what it should look like, that I think could not be fixed with the standard troubleshooting strategies.
>> >> Do you have any solutions for this? Is it for instance possible to
make changes to the recon-all script to shift the surfaces?
>> >> Thanks in advance, >> >> Doety > > _______________________________________________ Freesurfer mailing list Freesurfer@nmr.mgh.harvard.edu https://mail.nmr.mgh.harvard.edu/mailman/listinfo/freesurfer
The information in this e-mail is intended only for the person to whom
it is
addressed. If you believe this e-mail was sent to you in error and the
contains patient information, please contact the Partners Compliance
HelpLine at
http://www.partners.org/complianceline . If the e-mail was sent to you
in error
but does not contain patient information, please contact the sender
and properly
dispose of the e-mail.
Freesurfer mailing list Freesurfer@nmr.mgh.harvard.edu https://mail.nmr.mgh.harvard.edu/mailman/listinfo/freesurfer
Freesurfer mailing list Freesurfer@nmr.mgh.harvard.edu https://mail.nmr.mgh.harvard.edu/mailman/listinfo/freesurfer
Freesurfer mailing list Freesurfer@nmr.mgh.harvard.edu https://mail.nmr.mgh.harvard.edu/mailman/listinfo/freesurfer
Freesurfer mailing list Freesurfer@nmr.mgh.harvard.edu https://mail.nmr.mgh.harvard.edu/mailman/listinfo/freesurfer
Hi Bruce,
Thanks for your answer. I was trying to use the fsl segment in freesurfer indeed. But since I couldn't get that working, I am now trying to do the trick with mri_segment. Sorry to ask you again, but I still have troubles with mri_segment. So I am trying to use the -wm_low flag to make the program do a different segmentation than the default. But it looks like mri_segment is still using the default settings? And the wm.new.mgz I get out of it looks completely the same as my previous wm.mgz
So my command is: mri_segment brain.mgz wm.new.mgz -wlo 110
This is what comes out:
doing initial intensity segmentation... using local statistics to label ambiguous voxels... computing class statistics for intensity windows... WM (104.0): 105.4 +- 4.8 [80.0 --> 125.0] GM (70.0) : 68.4 +- 10.3 [30.0 --> 96.0] setting bottom of white matter range to 78.6 setting top of gray matter range to 88.9 doing initial intensity segmentation... using local statistics to label ambiguous voxels... using local geometry to label remaining ambiguous voxels...
reclassifying voxels using Gaussian border classifier...
removing voxels with positive offset direction... smoothing T1 volume with sigma = 0.250 removing 1-dimensional structures... 3683 sparsely connected voxels removed... thickening thin strands.... 20 segments, 2500 filled 8702 bright non-wm voxels segmented. 4341 diagonally connected voxels added... white matter segmentation took 1.5 minutes writing output to wm.new.mgz...
I put the output in bold which (I think) could be causing troubles(?) It looks like mri_segment is still using the default limits for wm and gm. I tried the same adding the flag -noauto, but it didn't make any difference.
Do you know what I am doing wrong here? How should I use mri_segment in order to really set different ranges of wm and gm intensities?
Again, thanks for your help!!
Doety
Hi Doety
for mri_segment the options have to come before the required arguments not at the end of the command line.
cheers Bruce On Wed, 16 Jul 2014, Doety Prins wrote:
Hi Bruce, Thanks for your answer. I was trying to use the fsl segment in freesurfer indeed. But since I couldn't get that working, I am now trying to do the trick with mri_segment. Sorry to ask you again, but I still have troubles with mri_segment. So I am trying to use the -wm_low flag to make the program do a different segmentation than the default. But it looks like mri_segment is still using the default settings? And the wm.new.mgz I get out of it looks completely the same as my previous wm.mgz
So my command is: mri_segment brain.mgz wm.new.mgz -wlo 110
This is what comes out:
doing initial intensity segmentation... using local statistics to label ambiguous voxels... computing class statistics for intensity windows... WM (104.0): 105.4 +- 4.8 [80.0 --> 125.0] GM (70.0) : 68.4 +- 10.3 [30.0 --> 96.0] setting bottom of white matter range to 78.6 setting top of gray matter range to 88.9 doing initial intensity segmentation... using local statistics to label ambiguous voxels... using local geometry to label remaining ambiguous voxels...
reclassifying voxels using Gaussian border classifier...
removing voxels with positive offset direction... smoothing T1 volume with sigma = 0.250 removing 1-dimensional structures... 3683 sparsely connected voxels removed... thickening thin strands.... 20 segments, 2500 filled 8702 bright non-wm voxels segmented. 4341 diagonally connected voxels added... white matter segmentation took 1.5 minutes writing output to wm.new.mgz...
I put the output in bold which (I think) could be causing troubles(?) It looks like mri_segment is still using the default limits for wm and gm. I tried the same adding the flag -noauto, but it didn't make any difference.
Do you know what I am doing wrong here? How should I use mri_segment in order to really set different ranges of wm and gm intensities?
Again, thanks for your help!!
Doety
glad to hear it Bruce On Thu, 17 Jul 2014, Doety Prins wrote:
Thanks Bruce! Now it's working, and the wm.mgz looks good!
Doety _______________________________________________ Freesurfer mailing list Freesurfer@nmr.mgh.harvard.edu https://mail.nmr.mgh.harvard.edu/mailman/listinfo/freesurfer
Just for the record, I accidentally e-mailed you that the slice thickness was 2 mm, I now found out that the slice thickness is just 1 mm.
Doety
freesurfer@nmr.mgh.harvard.edu
-
 Bruce Fischl
Bruce Fischl -
 Doety Prins
Doety Prins -
 Louis Nicholas Vinke
Louis Nicholas Vinke