
Hi Brian,
when you use the longitudinal stream in FreeSurfer, it accounts for the 'relatedness' therefore the resulting information (e.g. segmentation labels, volume measures, thickness measures, surface locations etc. ) will be more accurate (on average) than if you'd just process the different time points independently (cross sectionally).
Comparing the results after FreeSurfer is through will work exactly the same in both settings. You can compute what you are interested in (e.g. percent volume loss in left hippocampus by looking into the aseg.stats , or percent cortical thinning) per subject and then do a group analysis with your two groups.
Best, Martin
On Thu, 2009-10-01 at 14:39 -0400, Gogtay, Nitin (NIH/NIMH) [E] wrote:
Hi Martin + others,
Thank you for your help on this.
I am still a bit confused and maybe you can help me understand. So we have a sample of 12 patients + 12 controls and each with 2 scans (time 1 and time 2) thus total 48 scans.
As suggested these have been processed using the longitudinal free surfer algorithm (Thanks for your help regarding troubleshooting during that as well)
To compare the results between two time points (I assume slopes of subjects compared to those of controls? And intercepts?), how does one account for the 'relatedness' i.e. Same subject two time points? As it is now, wouldn't the free surfer treat the data as 'cross sectional' to calculate the slopes?
Maybe i/we are missing something?
Sorry for the bother and thank you again for the help!
Nitin
Nitin Gogtay, M.D. Staff Clinician Child Psychiatry Branch, NIMH (301) 435 4494
From: "Weisinger, Brian (NIH/OD) [E]" Brian.Weisinger@nih.gov Date: Thu, 1 Oct 2009 14:26:23 -0400 To: Nitin Gogtay gogtayn@mail.nih.gov Subject: FW: [Freesurfer] How to calculate differences using the longitudinal data
------ Forwarded Message From: Martin Reuter mreuter@nmr.mgh.harvard.edu Date: Wed, 30 Sep 2009 14:49:46 -0400 To: "Weisinger, Brian (NIH/OD) [E]" Brian.Weisinger@nih.gov Cc: "Freesurfer@nmr.mgh.harvard.edu" Freesurfer@nmr.mgh.harvard.edu Subject: Re: [Freesurfer] How to calculate differences using the longitudinal data
Hi Brian,
after the longitudinal runs are through you have directories tpN.long.base for each timepoint per subject. These contain the same structure as the cross sectionally processed results, however the values (e.g. thickness, volume etc. in the stat files) should be more accurate.
You can compare these results between timepoints in the same way you would do that without the longitudinal stream. You just need to take the more accurate information in the .long. directories instead.
Best, Martin
On Tue, 2009-09-29 at 14:59 -0400, Weisinger, Brian (NIH/OD) [E] wrote:
I have run the longitudinal stream on the scans I need, but now I am not sure what to use to compare the differences. If someone could clarify this for me I would really appreciate it.
Thanks
Freesurfer mailing list Freesurfer@nmr.mgh.harvard.edu https://mail.nmr.mgh.harvard.edu/mailman/listinfo/freesurfer
------ End of Forwarded Message

If you do a text search on our wiki for "paired" it will take you to the instructions for doing a paired analysis.
doug
Martin Reuter wrote:
Hi Brian,
when you use the longitudinal stream in FreeSurfer, it accounts for the 'relatedness' therefore the resulting information (e.g. segmentation labels, volume measures, thickness measures, surface locations etc. ) will be more accurate (on average) than if you'd just process the different time points independently (cross sectionally).
Comparing the results after FreeSurfer is through will work exactly the same in both settings. You can compute what you are interested in (e.g. percent volume loss in left hippocampus by looking into the aseg.stats , or percent cortical thinning) per subject and then do a group analysis with your two groups.
Best, Martin
On Thu, 2009-10-01 at 14:39 -0400, Gogtay, Nitin (NIH/NIMH) [E] wrote:
Hi Martin + others,
Thank you for your help on this.
I am still a bit confused and maybe you can help me understand. So we have a sample of 12 patients + 12 controls and each with 2 scans (time 1 and time 2) thus total 48 scans.
As suggested these have been processed using the longitudinal free surfer algorithm (Thanks for your help regarding troubleshooting during that as well)
To compare the results between two time points (I assume slopes of subjects compared to those of controls? And intercepts?), how does one account for the 'relatedness' i.e. Same subject two time points? As it is now, wouldn't the free surfer treat the data as 'cross sectional' to calculate the slopes?
Maybe i/we are missing something?
Sorry for the bother and thank you again for the help!
Nitin
Nitin Gogtay, M.D. Staff Clinician Child Psychiatry Branch, NIMH (301) 435 4494
From: "Weisinger, Brian (NIH/OD) [E]" Brian.Weisinger@nih.gov Date: Thu, 1 Oct 2009 14:26:23 -0400 To: Nitin Gogtay gogtayn@mail.nih.gov Subject: FW: [Freesurfer] How to calculate differences using the longitudinal data
------ Forwarded Message From: Martin Reuter mreuter@nmr.mgh.harvard.edu Date: Wed, 30 Sep 2009 14:49:46 -0400 To: "Weisinger, Brian (NIH/OD) [E]" Brian.Weisinger@nih.gov Cc: "Freesurfer@nmr.mgh.harvard.edu" Freesurfer@nmr.mgh.harvard.edu Subject: Re: [Freesurfer] How to calculate differences using the longitudinal data
Hi Brian,
after the longitudinal runs are through you have directories tpN.long.base for each timepoint per subject. These contain the same structure as the cross sectionally processed results, however the values (e.g. thickness, volume etc. in the stat files) should be more accurate.
You can compare these results between timepoints in the same way you would do that without the longitudinal stream. You just need to take the more accurate information in the .long. directories instead.
Best, Martin
On Tue, 2009-09-29 at 14:59 -0400, Weisinger, Brian (NIH/OD) [E] wrote:
I have run the longitudinal stream on the scans I need, but now I am not sure what to use to compare the differences. If someone could clarify this for me I would really appreciate it.
Thanks
Freesurfer mailing list Freesurfer@nmr.mgh.harvard.edu https://mail.nmr.mgh.harvard.edu/mailman/listinfo/freesurfer
------ End of Forwarded Message
Freesurfer mailing list Freesurfer@nmr.mgh.harvard.edu https://mail.nmr.mgh.harvard.edu/mailman/listinfo/freesurfer

I was running
recon-all -base Sub1 -tp 00308 -tp 01150 -all
After a couple of minutes it gave me this error message.....
Fri Oct 2 09:48:05 EDT 2009 talairach_avi done \n cp transforms/talairach.auto.xfm transforms/talairach.xfm \n #-------------------------------------------- #@# Talairach Failure Detection Fri Oct 2 09:48:06 EDT 2009 /Applications/freesurfer/subjects/Sub1/mri \n talairach_afd -T 0.005 -xfm transforms/talairach.xfm \n ERROR: talairach_afd: Talairach Transform: transforms/talairach.xfm ***FAILED*** (p=0.0000, pval=0.0000 < threshold=0.0050) Darwin CHP-MP2.local 9.8.0 Darwin Kernel Version 9.8.0: Wed Jul 15 16:55:01 PDT 2009; root:xnu-1228.15.4~1/RELEASE_I386 i386
recon-all exited with ERRORS at Fri Oct 2 09:48:06 EDT 2009
Any ideas how to fix this
On 10/1/09 3:12 PM, "Martin Reuter" mreuter@nmr.mgh.harvard.edu wrote:
Hi Brian,
when you use the longitudinal stream in FreeSurfer, it accounts for the 'relatedness' therefore the resulting information (e.g. segmentation labels, volume measures, thickness measures, surface locations etc. ) will be more accurate (on average) than if you'd just process the different time points independently (cross sectionally).
Comparing the results after FreeSurfer is through will work exactly the same in both settings. You can compute what you are interested in (e.g. percent volume loss in left hippocampus by looking into the aseg.stats , or percent cortical thinning) per subject and then do a group analysis with your two groups.
Best, Martin
On Thu, 2009-10-01 at 14:39 -0400, Gogtay, Nitin (NIH/NIMH) [E] wrote:
Hi Martin + others,
Thank you for your help on this.
I am still a bit confused and maybe you can help me understand. So we have a sample of 12 patients + 12 controls and each with 2 scans (time 1 and time 2) thus total 48 scans.
As suggested these have been processed using the longitudinal free surfer algorithm (Thanks for your help regarding troubleshooting during that as well)
To compare the results between two time points (I assume slopes of subjects compared to those of controls? And intercepts?), how does one account for the 'relatedness' i.e. Same subject two time points? As it is now, wouldn't the free surfer treat the data as 'cross sectional' to calculate the slopes?
Maybe i/we are missing something?
Sorry for the bother and thank you again for the help!
Nitin
Nitin Gogtay, M.D. Staff Clinician Child Psychiatry Branch, NIMH (301) 435 4494
From: "Weisinger, Brian (NIH/OD) [E]" Brian.Weisinger@nih.gov Date: Thu, 1 Oct 2009 14:26:23 -0400 To: Nitin Gogtay gogtayn@mail.nih.gov Subject: FW: [Freesurfer] How to calculate differences using the longitudinal data
------ Forwarded Message From: Martin Reuter mreuter@nmr.mgh.harvard.edu Date: Wed, 30 Sep 2009 14:49:46 -0400 To: "Weisinger, Brian (NIH/OD) [E]" Brian.Weisinger@nih.gov Cc: "Freesurfer@nmr.mgh.harvard.edu" Freesurfer@nmr.mgh.harvard.edu Subject: Re: [Freesurfer] How to calculate differences using the longitudinal data
Hi Brian,
after the longitudinal runs are through you have directories tpN.long.base for each timepoint per subject. These contain the same structure as the cross sectionally processed results, however the values (e.g. thickness, volume etc. in the stat files) should be more accurate.
You can compare these results between timepoints in the same way you would do that without the longitudinal stream. You just need to take the more accurate information in the .long. directories instead.
Best, Martin
On Tue, 2009-09-29 at 14:59 -0400, Weisinger, Brian (NIH/OD) [E] wrote:
I have run the longitudinal stream on the scans I need, but now I am not sure what to use to compare the differences. If someone could clarify this for me I would really appreciate it.
Thanks
Freesurfer mailing list Freesurfer@nmr.mgh.harvard.edu https://mail.nmr.mgh.harvard.edu/mailman/listinfo/freesurfer
------ End of Forwarded Message
Looks like the talairach check failed. Check the talairach.xfm (as you would do in the cross sectional processing, when this happens -> tutorial). If it looks OK try processing with -notal-check to skip the check. If it looks wrong, try to correct it with tkregister manually. Sometimes the transform is OK, but the check fails.
Best, Martin
On Fri, 2009-10-02 at 09:51 -0400, Weisinger, Brian (NIH/OD) [E] wrote:
I was running
recon-all -base Sub1 -tp 00308 -tp 01150 -all
After a couple of minutes it gave me this error message.....
Fri Oct 2 09:48:05 EDT 2009 talairach_avi done \n cp transforms/talairach.auto.xfm transforms/talairach.xfm \n #-------------------------------------------- #@# Talairach Failure Detection Fri Oct 2 09:48:06 EDT 2009 /Applications/freesurfer/subjects/Sub1/mri \n talairach_afd -T 0.005 -xfm transforms/talairach.xfm \n ERROR: talairach_afd: Talairach Transform: transforms/talairach.xfm ***FAILED*** (p=0.0000, pval=0.0000 < threshold=0.0050) Darwin CHP-MP2.local 9.8.0 Darwin Kernel Version 9.8.0: Wed Jul 15 16:55:01 PDT 2009; root:xnu-1228.15.4~1/RELEASE_I386 i386
recon-all exited with ERRORS at Fri Oct 2 09:48:06 EDT 2009
Any ideas how to fix this
On 10/1/09 3:12 PM, "Martin Reuter" mreuter@nmr.mgh.harvard.edu wrote:
Hi Brian,
when you use the longitudinal stream in FreeSurfer, it accounts for the 'relatedness' therefore the resulting information (e.g. segmentation labels, volume measures, thickness measures, surface locations etc. ) will be more accurate (on average) than if you'd just process the different time points independently (cross sectionally).
Comparing the results after FreeSurfer is through will work exactly the same in both settings. You can compute what you are interested in (e.g. percent volume loss in left hippocampus by looking into the aseg.stats , or percent cortical thinning) per subject and then do a group analysis with your two groups.
Best, Martin
On Thu, 2009-10-01 at 14:39 -0400, Gogtay, Nitin (NIH/NIMH) [E] wrote:
Hi Martin + others,
Thank you for your help on this.
I am still a bit confused and maybe you can help me understand. So we have a sample of 12 patients + 12 controls and each with 2 scans (time 1 and time 2) thus total 48 scans.
As suggested these have been processed using the longitudinal free surfer algorithm (Thanks for your help regarding troubleshooting during that as well)
To compare the results between two time points (I assume slopes of subjects compared to those of controls? And intercepts?), how does one account for the 'relatedness' i.e. Same subject two time points? As it is now, wouldn't the free surfer treat the data as 'cross sectional' to calculate the slopes?
Maybe i/we are missing something?
Sorry for the bother and thank you again for the help!
Nitin
Nitin Gogtay, M.D. Staff Clinician Child Psychiatry Branch, NIMH (301) 435 4494
From: "Weisinger, Brian (NIH/OD) [E]" Brian.Weisinger@nih.gov Date: Thu, 1 Oct 2009 14:26:23 -0400 To: Nitin Gogtay gogtayn@mail.nih.gov Subject: FW: [Freesurfer] How to calculate differences using the longitudinal data
------ Forwarded Message From: Martin Reuter mreuter@nmr.mgh.harvard.edu Date: Wed, 30 Sep 2009 14:49:46 -0400 To: "Weisinger, Brian (NIH/OD) [E]" Brian.Weisinger@nih.gov Cc: "Freesurfer@nmr.mgh.harvard.edu" Freesurfer@nmr.mgh.harvard.edu Subject: Re: [Freesurfer] How to calculate differences using the longitudinal data
Hi Brian,
after the longitudinal runs are through you have directories tpN.long.base for each timepoint per subject. These contain the same structure as the cross sectionally processed results, however the values (e.g. thickness, volume etc. in the stat files) should be more accurate.
You can compare these results between timepoints in the same way you would do that without the longitudinal stream. You just need to take the more accurate information in the .long. directories instead.
Best, Martin
On Tue, 2009-09-29 at 14:59 -0400, Weisinger, Brian (NIH/OD) [E] wrote:
I have run the longitudinal stream on the scans I need, but now I am not sure what to use to compare the differences. If someone could clarify this for me I would really appreciate it.
Thanks
Freesurfer mailing list Freesurfer@nmr.mgh.harvard.edu https://mail.nmr.mgh.harvard.edu/mailman/listinfo/freesurfer
------ End of Forwarded Message
How exactly do I check the talairach.xfm? Are there instructions somewhere? I looked at the subjects brains with tkmedit, tksurfer and they look fine.
On 10/2/09 1:15 PM, "Martin Reuter" mreuter@nmr.mgh.harvard.edu wrote:
Looks like the talairach check failed. Check the talairach.xfm (as you would do in the cross sectional processing, when this happens -> tutorial). If it looks OK try processing with -notal-check to skip the check. If it looks wrong, try to correct it with tkregister manually. Sometimes the transform is OK, but the check fails.
Best, Martin
On Fri, 2009-10-02 at 09:51 -0400, Weisinger, Brian (NIH/OD) [E] wrote:
I was running
recon-all -base Sub1 -tp 00308 -tp 01150 -all
After a couple of minutes it gave me this error message.....
Fri Oct 2 09:48:05 EDT 2009 talairach_avi done \n cp transforms/talairach.auto.xfm transforms/talairach.xfm \n #-------------------------------------------- #@# Talairach Failure Detection Fri Oct 2 09:48:06 EDT 2009 /Applications/freesurfer/subjects/Sub1/mri \n talairach_afd -T 0.005 -xfm transforms/talairach.xfm \n ERROR: talairach_afd: Talairach Transform: transforms/talairach.xfm ***FAILED*** (p=0.0000, pval=0.0000 < threshold=0.0050) Darwin CHP-MP2.local 9.8.0 Darwin Kernel Version 9.8.0: Wed Jul 15 16:55:01 PDT 2009; root:xnu-1228.15.4~1/RELEASE_I386 i386
recon-all exited with ERRORS at Fri Oct 2 09:48:06 EDT 2009
Any ideas how to fix this
On 10/1/09 3:12 PM, "Martin Reuter" mreuter@nmr.mgh.harvard.edu wrote:
Hi Brian,
when you use the longitudinal stream in FreeSurfer, it accounts for the 'relatedness' therefore the resulting information (e.g. segmentation labels, volume measures, thickness measures, surface locations etc. ) will be more accurate (on average) than if you'd just process the different time points independently (cross sectionally).
Comparing the results after FreeSurfer is through will work exactly the same in both settings. You can compute what you are interested in (e.g. percent volume loss in left hippocampus by looking into the aseg.stats , or percent cortical thinning) per subject and then do a group analysis with your two groups.
Best, Martin
On Thu, 2009-10-01 at 14:39 -0400, Gogtay, Nitin (NIH/NIMH) [E] wrote:
Hi Martin + others,
Thank you for your help on this.
I am still a bit confused and maybe you can help me understand. So we have a sample of 12 patients + 12 controls and each with 2 scans (time 1 and time 2) thus total 48 scans.
As suggested these have been processed using the longitudinal free surfer algorithm (Thanks for your help regarding troubleshooting during that as well)
To compare the results between two time points (I assume slopes of subjects compared to those of controls? And intercepts?), how does one account for the 'relatedness' i.e. Same subject two time points? As it is now, wouldn't the free surfer treat the data as 'cross sectional' to calculate the slopes?
Maybe i/we are missing something?
Sorry for the bother and thank you again for the help!
Nitin
Nitin Gogtay, M.D. Staff Clinician Child Psychiatry Branch, NIMH (301) 435 4494
From: "Weisinger, Brian (NIH/OD) [E]" Brian.Weisinger@nih.gov Date: Thu, 1 Oct 2009 14:26:23 -0400 To: Nitin Gogtay gogtayn@mail.nih.gov Subject: FW: [Freesurfer] How to calculate differences using the longitudinal data
------ Forwarded Message From: Martin Reuter mreuter@nmr.mgh.harvard.edu Date: Wed, 30 Sep 2009 14:49:46 -0400 To: "Weisinger, Brian (NIH/OD) [E]" Brian.Weisinger@nih.gov Cc: "Freesurfer@nmr.mgh.harvard.edu" Freesurfer@nmr.mgh.harvard.edu Subject: Re: [Freesurfer] How to calculate differences using the longitudinal data
Hi Brian,
after the longitudinal runs are through you have directories tpN.long.base for each timepoint per subject. These contain the same structure as the cross sectionally processed results, however the values (e.g. thickness, volume etc. in the stat files) should be more accurate.
You can compare these results between timepoints in the same way you would do that without the longitudinal stream. You just need to take the more accurate information in the .long. directories instead.
Best, Martin
On Tue, 2009-09-29 at 14:59 -0400, Weisinger, Brian (NIH/OD) [E] wrote:
I have run the longitudinal stream on the scans I need, but now I am not sure what to use to compare the differences. If someone could clarify this for me I would really appreciate it.
Thanks
Freesurfer mailing list Freesurfer@nmr.mgh.harvard.edu https://mail.nmr.mgh.harvard.edu/mailman/listinfo/freesurfer
------ End of Forwarded Message
When you said to process it with the -notal-check to skip the talairach check....am I supposed to add that in the first recon all or add it to the recon all base command?
On 10/2/09 1:15 PM, "Martin Reuter" mreuter@nmr.mgh.harvard.edu wrote:
Looks like the talairach check failed. Check the talairach.xfm (as you would do in the cross sectional processing, when this happens -> tutorial). If it looks OK try processing with -notal-check to skip the check. If it looks wrong, try to correct it with tkregister manually. Sometimes the transform is OK, but the check fails.
Best, Martin
On Fri, 2009-10-02 at 09:51 -0400, Weisinger, Brian (NIH/OD) [E] wrote:
I was running
recon-all -base Sub1 -tp 00308 -tp 01150 -all
After a couple of minutes it gave me this error message.....
Fri Oct 2 09:48:05 EDT 2009 talairach_avi done \n cp transforms/talairach.auto.xfm transforms/talairach.xfm \n #-------------------------------------------- #@# Talairach Failure Detection Fri Oct 2 09:48:06 EDT 2009 /Applications/freesurfer/subjects/Sub1/mri \n talairach_afd -T 0.005 -xfm transforms/talairach.xfm \n ERROR: talairach_afd: Talairach Transform: transforms/talairach.xfm ***FAILED*** (p=0.0000, pval=0.0000 < threshold=0.0050) Darwin CHP-MP2.local 9.8.0 Darwin Kernel Version 9.8.0: Wed Jul 15 16:55:01 PDT 2009; root:xnu-1228.15.4~1/RELEASE_I386 i386
recon-all exited with ERRORS at Fri Oct 2 09:48:06 EDT 2009
Any ideas how to fix this
On 10/1/09 3:12 PM, "Martin Reuter" mreuter@nmr.mgh.harvard.edu wrote:
Hi Brian,
when you use the longitudinal stream in FreeSurfer, it accounts for the 'relatedness' therefore the resulting information (e.g. segmentation labels, volume measures, thickness measures, surface locations etc. ) will be more accurate (on average) than if you'd just process the different time points independently (cross sectionally).
Comparing the results after FreeSurfer is through will work exactly the same in both settings. You can compute what you are interested in (e.g. percent volume loss in left hippocampus by looking into the aseg.stats , or percent cortical thinning) per subject and then do a group analysis with your two groups.
Best, Martin
On Thu, 2009-10-01 at 14:39 -0400, Gogtay, Nitin (NIH/NIMH) [E] wrote:
Hi Martin + others,
Thank you for your help on this.
I am still a bit confused and maybe you can help me understand. So we have a sample of 12 patients + 12 controls and each with 2 scans (time 1 and time 2) thus total 48 scans.
As suggested these have been processed using the longitudinal free surfer algorithm (Thanks for your help regarding troubleshooting during that as well)
To compare the results between two time points (I assume slopes of subjects compared to those of controls? And intercepts?), how does one account for the 'relatedness' i.e. Same subject two time points? As it is now, wouldn't the free surfer treat the data as 'cross sectional' to calculate the slopes?
Maybe i/we are missing something?
Sorry for the bother and thank you again for the help!
Nitin
Nitin Gogtay, M.D. Staff Clinician Child Psychiatry Branch, NIMH (301) 435 4494
From: "Weisinger, Brian (NIH/OD) [E]" Brian.Weisinger@nih.gov Date: Thu, 1 Oct 2009 14:26:23 -0400 To: Nitin Gogtay gogtayn@mail.nih.gov Subject: FW: [Freesurfer] How to calculate differences using the longitudinal data
------ Forwarded Message From: Martin Reuter mreuter@nmr.mgh.harvard.edu Date: Wed, 30 Sep 2009 14:49:46 -0400 To: "Weisinger, Brian (NIH/OD) [E]" Brian.Weisinger@nih.gov Cc: "Freesurfer@nmr.mgh.harvard.edu" Freesurfer@nmr.mgh.harvard.edu Subject: Re: [Freesurfer] How to calculate differences using the longitudinal data
Hi Brian,
after the longitudinal runs are through you have directories tpN.long.base for each timepoint per subject. These contain the same structure as the cross sectionally processed results, however the values (e.g. thickness, volume etc. in the stat files) should be more accurate.
You can compare these results between timepoints in the same way you would do that without the longitudinal stream. You just need to take the more accurate information in the .long. directories instead.
Best, Martin
On Tue, 2009-09-29 at 14:59 -0400, Weisinger, Brian (NIH/OD) [E] wrote:
I have run the longitudinal stream on the scans I need, but now I am not sure what to use to compare the differences. If someone could clarify this for me I would really appreciate it.
Thanks
Freesurfer mailing list Freesurfer@nmr.mgh.harvard.edu https://mail.nmr.mgh.harvard.edu/mailman/listinfo/freesurfer
------ End of Forwarded Message
Only to the runs, where the check fails (if the transform looks OK). So in your case the -base run.
Good luck, Martin
On Mon, 2009-10-05 at 10:01 -0400, Weisinger, Brian (NIH/OD) [E] wrote:
When you said to process it with the -notal-check to skip the talairach check....am I supposed to add that in the first recon all or add it to the recon all base command?
On 10/2/09 1:15 PM, "Martin Reuter" mreuter@nmr.mgh.harvard.edu wrote:
Looks like the talairach check failed. Check the talairach.xfm (as you would do in the cross sectional processing, when this happens -> tutorial). If it looks OK try processing with -notal-check to skip the check. If it looks wrong, try to correct it with tkregister manually. Sometimes the transform is OK, but the check fails.
Best, Martin
On Fri, 2009-10-02 at 09:51 -0400, Weisinger, Brian (NIH/OD) [E] wrote:
I was running
recon-all -base Sub1 -tp 00308 -tp 01150 -all
After a couple of minutes it gave me this error message.....
Fri Oct 2 09:48:05 EDT 2009 talairach_avi done \n cp transforms/talairach.auto.xfm transforms/talairach.xfm \n #-------------------------------------------- #@# Talairach Failure Detection Fri Oct 2 09:48:06 EDT 2009 /Applications/freesurfer/subjects/Sub1/mri \n talairach_afd -T 0.005 -xfm transforms/talairach.xfm \n ERROR: talairach_afd: Talairach Transform: transforms/talairach.xfm ***FAILED*** (p=0.0000, pval=0.0000 < threshold=0.0050) Darwin CHP-MP2.local 9.8.0 Darwin Kernel Version 9.8.0: Wed Jul 15 16:55:01 PDT 2009; root:xnu-1228.15.4~1/RELEASE_I386 i386
recon-all exited with ERRORS at Fri Oct 2 09:48:06 EDT 2009
Any ideas how to fix this
On 10/1/09 3:12 PM, "Martin Reuter" mreuter@nmr.mgh.harvard.edu wrote:
Hi Brian,
when you use the longitudinal stream in FreeSurfer, it accounts for the 'relatedness' therefore the resulting information (e.g. segmentation labels, volume measures, thickness measures, surface locations etc. ) will be more accurate (on average) than if you'd just process the different time points independently (cross sectionally).
Comparing the results after FreeSurfer is through will work exactly the same in both settings. You can compute what you are interested in (e.g. percent volume loss in left hippocampus by looking into the aseg.stats , or percent cortical thinning) per subject and then do a group analysis with your two groups.
Best, Martin
On Thu, 2009-10-01 at 14:39 -0400, Gogtay, Nitin (NIH/NIMH) [E] wrote:
Hi Martin + others,
Thank you for your help on this.
I am still a bit confused and maybe you can help me understand. So we have a sample of 12 patients + 12 controls and each with 2 scans (time 1 and time 2) thus total 48 scans.
As suggested these have been processed using the longitudinal free surfer algorithm (Thanks for your help regarding troubleshooting during that as well)
To compare the results between two time points (I assume slopes of subjects compared to those of controls? And intercepts?), how does one account for the 'relatedness' i.e. Same subject two time points? As it is now, wouldn't the free surfer treat the data as 'cross sectional' to calculate the slopes?
Maybe i/we are missing something?
Sorry for the bother and thank you again for the help!
Nitin
Nitin Gogtay, M.D. Staff Clinician Child Psychiatry Branch, NIMH (301) 435 4494
From: "Weisinger, Brian (NIH/OD) [E]" Brian.Weisinger@nih.gov Date: Thu, 1 Oct 2009 14:26:23 -0400 To: Nitin Gogtay gogtayn@mail.nih.gov Subject: FW: [Freesurfer] How to calculate differences using the longitudinal data
------ Forwarded Message From: Martin Reuter mreuter@nmr.mgh.harvard.edu Date: Wed, 30 Sep 2009 14:49:46 -0400 To: "Weisinger, Brian (NIH/OD) [E]" Brian.Weisinger@nih.gov Cc: "Freesurfer@nmr.mgh.harvard.edu" Freesurfer@nmr.mgh.harvard.edu Subject: Re: [Freesurfer] How to calculate differences using the longitudinal data
Hi Brian,
after the longitudinal runs are through you have directories tpN.long.base for each timepoint per subject. These contain the same structure as the cross sectionally processed results, however the values (e.g. thickness, volume etc. in the stat files) should be more accurate.
You can compare these results between timepoints in the same way you would do that without the longitudinal stream. You just need to take the more accurate information in the .long. directories instead.
Best, Martin
On Tue, 2009-09-29 at 14:59 -0400, Weisinger, Brian (NIH/OD) [E] wrote:
I have run the longitudinal stream on the scans I need, but now I am not sure what to use to compare the differences. If someone could clarify this for me I would really appreciate it.
Thanks
Freesurfer mailing list Freesurfer@nmr.mgh.harvard.edu https://mail.nmr.mgh.harvard.edu/mailman/listinfo/freesurfer
------ End of Forwarded Message
When I try to use tkregister2 I get this message, how do I go about changing not being able to save the changes I make.
WARNING: cannot write to /Applications/freesurfer/subjects/00007/mri/transforms. You will not be able to save any edits. Hit Enter to continue:
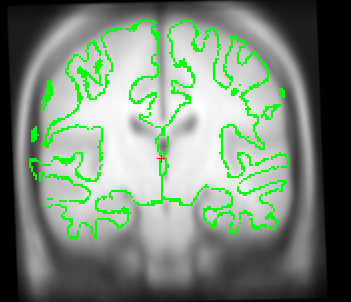
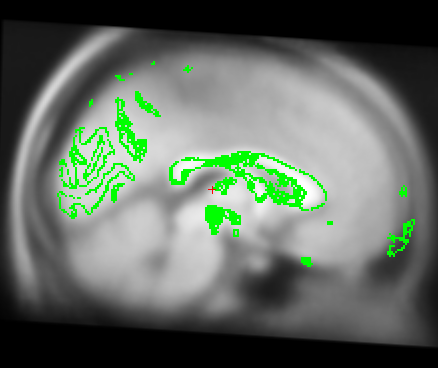
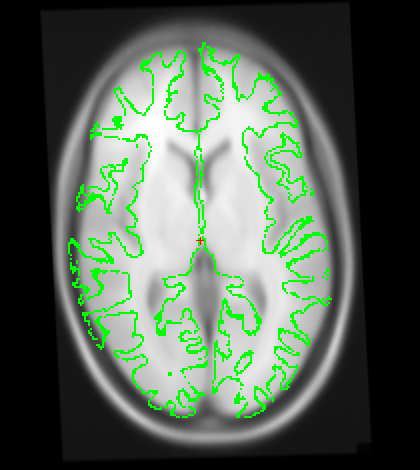
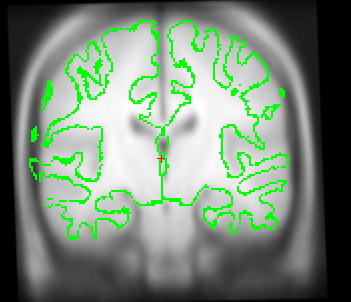
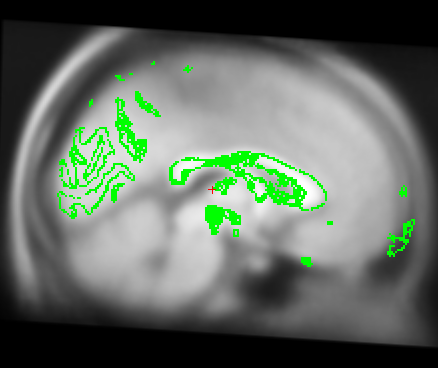
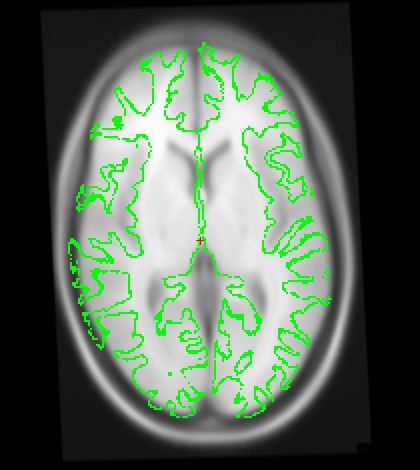
Also I just wanted to get your opinion about these images...It looks like I need to stretch them some so that the green part is in the white brain area. Would you agree?
[cid:3337666238_142370727]
[cid:3337666238_142392223]
[cid:3337666238_142372460]
{kind=link}
{kind=link}
{kind=link}
It means that you don't have permission to write into that directory.
The images look a little off. What kind of scanner are you using?
doug
Weisinger, Brian (NIH/OD) [E] wrote:
When I try to use tkregister2 I get this message, how do I go about changing not being able to save the changes I make.
WARNING: cannot write to /Applications/freesurfer/subjects/00007/mri/transforms. You will not be able to save any edits. Hit Enter to continue:
Also I just wanted to get your opinion about these images...It looks like I need to stretch them some so that the green part is in the white brain area. Would you agree?
[cid:3337666238_142370727]
[cid:3337666238_142392223]
[cid:3337666238_142372460]
Freesurfer mailing list Freesurfer@nmr.mgh.harvard.edu https://mail.nmr.mgh.harvard.edu/mailman/listinfo/freesurfer
Hi Martin,
I just wanted to get your opinion about these images...It looks like I need to stretch them some so that the green part is in the white brain area. Would you agree?
[cid:3337684698_143483721]
[cid:3337684698_143480418]
[cid:3337684698_143486052] ------ End of Forwarded Message
{kind=link}
{kind=link}
{kind=link}
Hi Brian,
yes it looks like this is slightly too small. A little stretching does not hurt. The talairach transform from the base is used for all the longitudinal runs, so if it is wrong, this will have an influence. In your case, however, I think it might not be a big problem (as the difference is really small). You could try both ways (with manual adjustment and without on one or two subjects) and see if it has a big influence. I would be surprised if it did.
Best, Martin
On Tue, 2009-10-06 at 14:38 -0400, Weisinger, Brian (NIH/OD) [E] wrote:
Hi Martin,
I just wanted to get your opinion about these images...It looks like I need to stretch them some so that the green part is in the white brain area. Would you agree?
[cid:3337684698_143483721]
[cid:3337684698_143480418]
[cid:3337684698_143486052] ------ End of Forwarded Message
Hi Brian,
coming back to your talairach.xfm question. Actually it is not really used for anything except the eTIV computation (estimated total intracranial volume). Therefore it is not really necessary to correct this unless you need that value. The talairach.lta however is used for the segmentation and copied from the base to each timepoint (concatenation of the transforms), therefore it should be as accurate as possible in the base run.
We are currently looking at your other bug. The base template is not created correctly, not sure why.
Best, Martin
On Tue, 2009-10-06 at 14:38 -0400, Weisinger, Brian (NIH/OD) [E] wrote:
Hi Martin,
I just wanted to get your opinion about these images...It looks like I need to stretch them some so that the green part is in the white brain area. Would you agree?
[cid:3337684698_143483721]
[cid:3337684698_143480418]
[cid:3337684698_143486052] ------ End of Forwarded Message

Hi everyone,
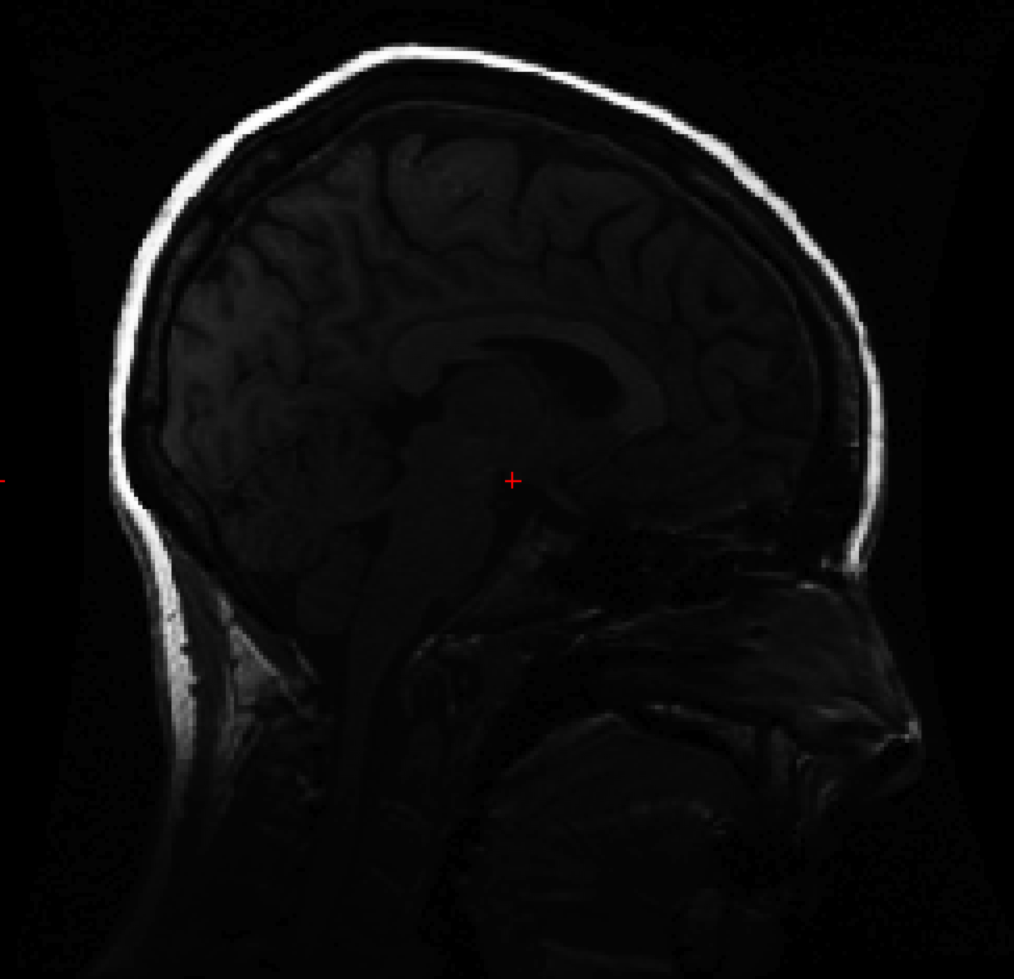
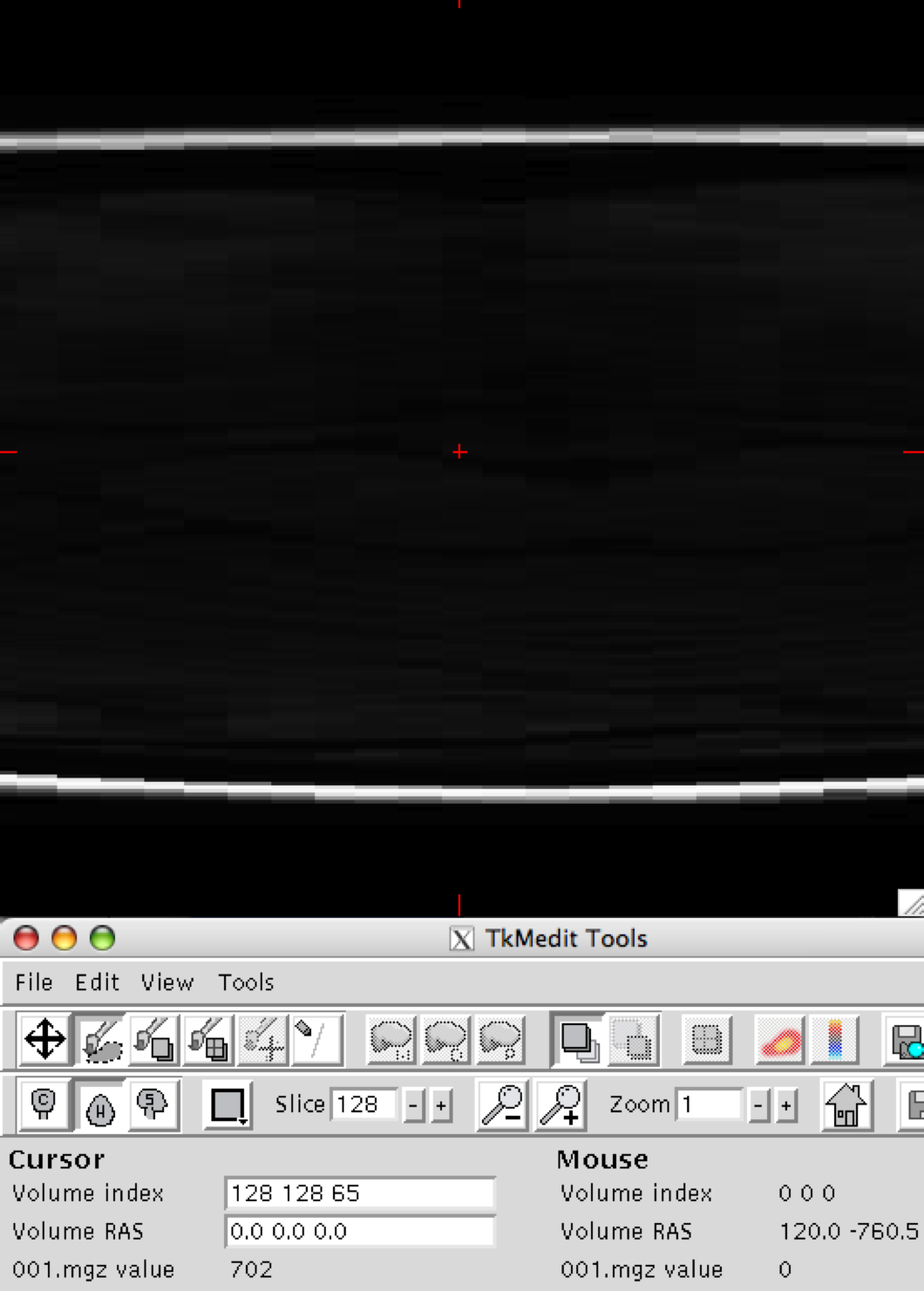
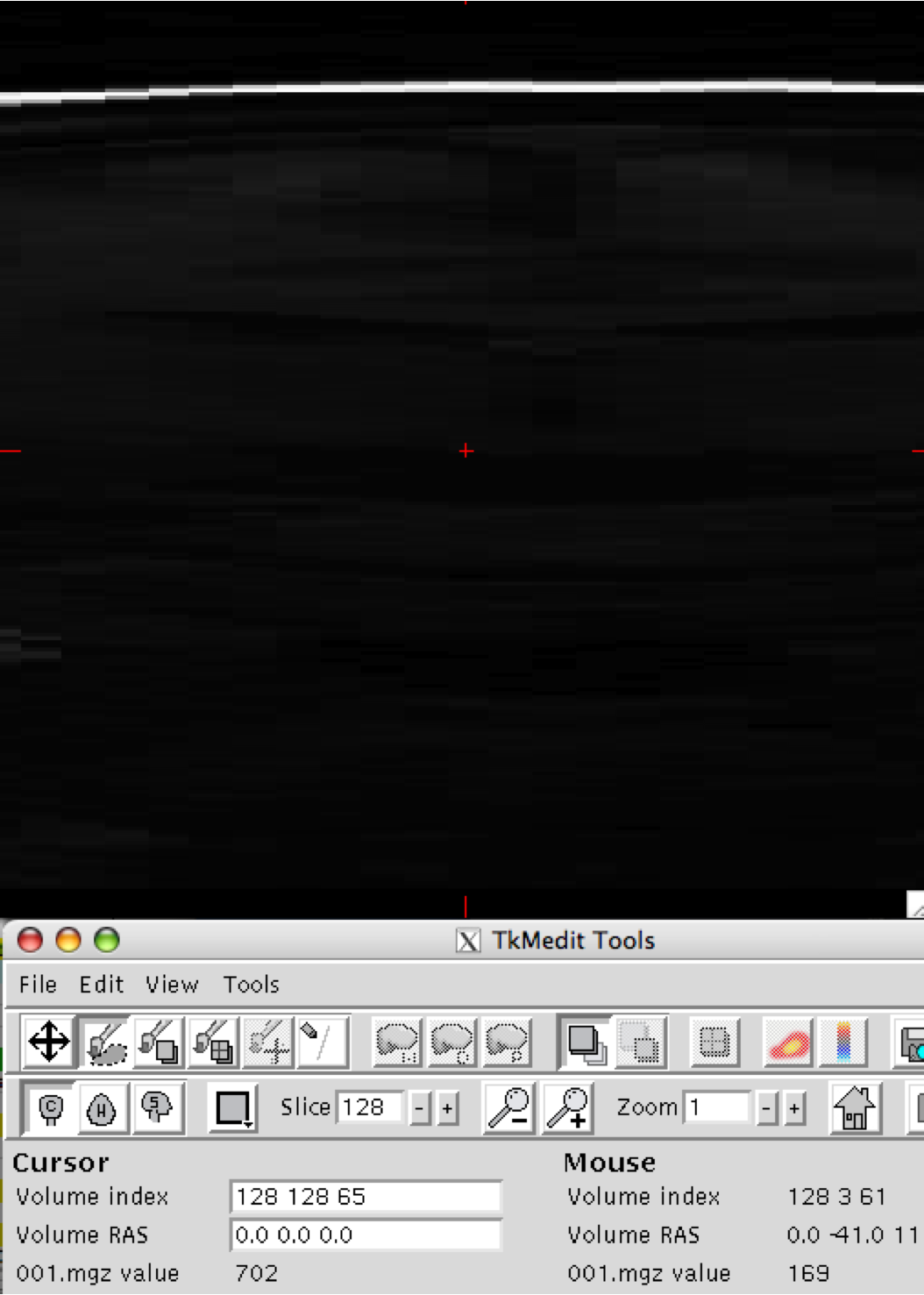
Thank you in advance for your help with this issue. When opening a subject's image with tkmedit to ensure proper orientation (after conversion to mgz format with recon-all), the sagittal view looks normal however the axial and coronal views show nothing but blur, for lack of a better word.
I have attached snapshots of all 3 planes. The original img files show no abnormalities. We searched the wiki, but were unable to find mention of this problem. I apologize if this has come up before.
We are curious to see if anyone has encountered this problem, and if there is a possible solution.
Thank you for your time and help,
Matt
{kind=link}
{kind=link}
{kind=link}
freesurfer@nmr.mgh.harvard.edu
-
 Douglas N Greve
Douglas N Greve -
 Martin Reuter
Martin Reuter -
 Warner, Matthew A.
Warner, Matthew A. -
 Weisinger, Brian (NIH/OD) [E]
Weisinger, Brian (NIH/OD) [E]