
Hi! Is there more documentation on what settings the mprage flag enables in recon-all? Specifically, I would be interested which individual steps it affects and what the parameters are for each of the steps. Thanks! Caspar

Hi Caspar
it changes some parameters to the intensity normalization and segmentation to account for the lower SNR and increased CNR in the mprage. You'll need to look in the recon-all script for the details
cheers Bruce
On Thu, 30 May 2013, Caspar M. Schwiedrzik wrote:
Hi! Is there more documentation on what settings the mprage flag enables in recon-all? Specifically, I would be interested which individual steps it affects and what the parameters are for each of the steps. Thanks! Caspar
I only find flags for mri_normalize and mri_segment in recon-all, and I have trouble reading the contents mri_normalize with a regular text editor. caspar
2013/5/31 Bruce Fischl fischl@nmr.mgh.harvard.edu
Hi Caspar
it changes some parameters to the intensity normalization and segmentation to account for the lower SNR and increased CNR in the mprage. You'll need to look in the recon-all script for the details
cheers Bruce
On Thu, 30 May 2013, Caspar M. Schwiedrzik wrote:
Hi!
Is there more documentation on what settings the mprage flag enables in recon-all? Specifically, I would be interested which individual steps it affects and what the parameters are for each of the steps. Thanks! Caspar
The information in this e-mail is intended only for the person to whom it is addressed. If you believe this e-mail was sent to you in error and the e-mail contains patient information, please contact the Partners Compliance HelpLine at http://www.partners.org/**compliancelinehttp://www.partners.org/complianceline. If the e-mail was sent to you in error but does not contain patient information, please contact the sender and properly dispose of the e-mail.

Hi Bruce and others,
We started a rather extensive push of data through the linux 5.2 release of free surfer. We also use OS X at times and I was hoping to get a R5.2 for MAC but I see it's not available. One of the guys that works in my lab — Jon Holt — also said that 5.2 was no longer available and indicated he heard there may be issues with 5.2
So two questions:
1) we have > 100 subjects pushed through R5.2 and still working inside 5.2, are we in trouble? What's the advice. We have many FTE hours on the editing and would be very painful to go back (we already did with with 5.0 to 5.2 change).
2) are we safe using 5.3 in conjunction with 5.2?
Any other advice about versions etc. Given the amount of time it takes to push data through it is difficult to keep changing versions, plus from a publication view it would be a hard sale I think to reviewers to indicate we used multiple versions.
Thanks,
-Robert
--------------------------------- Robert C. Welsh, Ph.D. Assistant Professor Department of Radiology Department of Psychiatry University of Michigan
********************************************************** Electronic Mail is not secure, may not be read every day, and should not be used for urgent or sensitive issues
Hi Robert
I'm really sorry to hear it. I suspect that though that you would be better off rerunning everything through 5.3. It should be the case that all manual edits are retained and it's just compute time that you will have wasted. 5.2 had a user-requested mod that made the surfaces inaccurate in some acquisitions, although not in our test suite, which of course is why we didn't catch it. We have announced the problem with 5.2 so I would worry that if you try to publish with it you will get yelled at by some reviewer.
Again, I'm sorry for the wasted time. We try hard to avoid this kind of problem, but our resources are quite finite and there's only so much testing we can do (and in this case, 5.2 passed all our internal tests).
Bruce
On Fri, 31 May 2013, Welsh, Robert wrote:
Hi Bruce and others,
We started a rather extensive push of data through the linux 5.2 release of free surfer. We also use OS X at times and I was hoping to get a R5.2 for MAC but I see it's not available. One of the guys that works in my lab — Jon Holt — also said that 5.2 was no longer available and indicated he heard there may be issues with 5.2
So two questions:
- we have > 100 subjects pushed through R5.2 and still working inside 5.2,
are we in trouble? What's the advice. We have many FTE hours on the editing and would be very painful to go back (we already did with with 5.0 to 5.2 change).
- are we safe using 5.3 in conjunction with 5.2?
Any other advice about versions etc. Given the amount of time it takes to push data through it is difficult to keep changing versions, plus from a publication view it would be a hard sale I think to reviewers to indicate we used multiple versions.
Thanks,
-Robert
Robert C. Welsh, Ph.D. Assistant Professor Department of Radiology Department of Psychiatry University of Michigan
Electronic Mail is not secure, may not be read every day, and should not be used for urgent or sensitive issues
mri_normalize is a binary so you can't, just recon-all.
On Fri, 31 May 2013, Caspar M. Schwiedrzik wrote:
I only find flags for mri_normalize and mri_segment in recon-all, and I have trouble reading the contents mri_normalize with a regular text editor. caspar
2013/5/31 Bruce Fischl fischl@nmr.mgh.harvard.edu Hi Caspar
it changes some parameters to the intensity normalization and segmentation to account for the lower SNR and increased CNR in the mprage. You'll need to look in the recon-all script for the details cheers Bruce On Thu, 30 May 2013, Caspar M. Schwiedrzik wrote: Hi! Is there more documentation on what settings the mprage flag enables in recon-all? Specifically, I would be interested which individual steps it affects and what the parameters are for each of the steps. Thanks! CasparThe information in this e-mail is intended only for the person to whom it is addressed. If you believe this e-mail was sent to you in error and the e-mail contains patient information, please contact the Partners Compliance HelpLine at http://www.partners.org/complianceline . If the e-mail was sent to you in error but does not contain patient information, please contact the sender and properly dispose of the e-mail.

Dear FSexperts,
the overlay scale bar consists of a colored and a gray part. Unfortunately, the gray in the binary colored represetation of the brain curvature is exactly the same as in the overlay scale bar. Is it possible to change the color in the scale bar background to white?
Regards Joerg
On Fri, 31 May 2013 16:20:24 -0400 (EDT) Bruce Fischl fischl@nmr.mgh.harvard.edu wrote:
mri_normalize is a binary so you can't, just recon-all.
On Fri, 31 May 2013, Caspar M. Schwiedrzik wrote:
I only find flags for mri_normalize and mri_segment in recon-all, and I have trouble reading the contents mri_normalize with a regular text editor. caspar
2013/5/31 Bruce Fischl fischl@nmr.mgh.harvard.edu Hi Caspar
it changes some parameters to the intensity normalization and segmentation to account for the lower SNR and increased CNR in the mprage. You'll need to look in the recon-all script for the details cheers Bruce On Thu, 30 May 2013, Caspar M. Schwiedrzik wrote: Hi! Is there more documentation on what settings the mprage flag enables in recon-all? Specifically, I would be interested which individual steps it affects and what the parameters are for each of the steps. Thanks! CasparThe information in this e-mail is intended only for the person to whom it is addressed. If you believe this e-mail was sent to you in error and the e-mail contains patient information, please contact the Partners Compliance HelpLine at http://www.partners.org/complianceline . If the e-mail was sent to you in error but does not contain patient information, please contact the sender and properly dispose of the e-mail.
you can change it to white via:
set scalebar_bright 255
cheers, Bruce
On Mon, 10 Jun 2013, Jörg Pfannmöller wrote:
Dear FSexperts,
the overlay scale bar consists of a colored and a gray part.
Unfortunately, the gray in the binary colored represetation of the brain curvature is exactly the same as in the overlay scale bar. Is it possible to change the color in the scale bar background to white?
Regards Joerg
On Fri, 31 May 2013 16:20:24 -0400 (EDT) Bruce Fischl fischl@nmr.mgh.harvard.edu wrote:
mri_normalize is a binary so you can't, just recon-all.
On Fri, 31 May 2013, Caspar M. Schwiedrzik wrote:
I only find flags for mri_normalize and mri_segment in recon-all, and I have trouble reading the contents mri_normalize with a regular text editor. caspar
2013/5/31 Bruce Fischl fischl@nmr.mgh.harvard.edu Hi Caspar
it changes some parameters to the intensity normalization and segmentation to account for the lower SNR and increased CNR in the mprage. You'll need to look in the recon-all script for the details cheers Bruce On Thu, 30 May 2013, Caspar M. Schwiedrzik wrote: Hi! Is there more documentation on what settings the mprage flag enables in recon-all? Specifically, I would be interested which individual steps it affects and what the parameters are for each of the steps. Thanks! CasparThe information in this e-mail is intended only for the person to whom it is addressed. If you believe this e-mail was sent to you in error and the e-mail contains patient information, please contact the Partners Compliance HelpLine at http://www.partners.org/complianceline . If the e-mail was sent to you in error but does not contain patient information, please contact the sender and properly dispose of the e-mail.
Hello FSexperts,
I am investigating a small area on the cortex. If this area is diplayed by tksurfer the lable outlines and the transition in the colors indicating the curvature looks rather low resolved. Probably this just shows the resolution of the fsaverage brain lattice. Is there a way to improve this and to make it look more smooth without smoothing the data? With other words, is it possible to artificially increase the resolution?
Regards Joerg
Hi Joerg
I think Jon Polimeni (ccd) might have some tools for this, but it's not something we routinely do
cheers Bruce
On Tue, 11 Jun 2013, Jörg Pfannmöller wrote:
Hello FSexperts,
I am investigating a small area on the cortex. If this area is diplayed by tksurfer the lable outlines and the transition in the colors indicating the curvature looks rather low resolved. Probably this just shows the resolution of the fsaverage brain lattice. Is there a way to improve this and to make it look more smooth without smoothing the data? With other words, is it possible to artificially increase the resolution?
Regards Joerg
Hello FSexperts,
I used the mgh memprage and the dura correction scheme described in:
http://www.mail-archive.com/freesurfer@nmr.mgh.harvard.edu/msg28479.html .
Afterwards I applied
recon-all -autorecon-pial -subjid subj_name
to the subjects in order to include the changes in the subsequent FS-Fast analysis. Currently I do not see any effect in tksurfer if I proceed like this. Please correct me if I am wrong with my effect size expectation. For completeness I add the command pipeline used:
recon-all -s subj_name -i /.../dicom_images.dcm recon-all -s subj_name -use-gpu -mprage -all mri_convert -odt float ./... .dcm $SUBJECTS_DIR/subj_name/mri/001.mgz mris_make_surfaces -dura 001.mgz 4 subj_name lh mris_make_surfaces -dura 001.mgz 4 subj_name rh recon-all -autorecon-pial -subjid subj_name
Hi Joerg
you need to convert the individual echoes to be in conformed space without scaling the intensities, then run it with something like
mris_make_surfaces -dura echo%d.mgz 4 subj_name lh
otherwise it will just load 001.mgz 4 times and think it is the different echoes. That is, name the echoes echo0.mgz echo1.mgz echo2.mgz and echo3.mgz
cheers Bruce
On Wed, 12 Jun 2013, Joerg Pfannmoeller wrote:
Hello FSexperts,
I used the mgh memprage and the dura correction scheme described in:
http://www.mail-archive.com/freesurfer@nmr.mgh.harvard.edu/msg28479.html .
Afterwards I applied
recon-all -autorecon-pial -subjid subj_name
to the subjects in order to include the changes in the subsequent FS-Fast analysis. Currently I do not see any effect in tksurfer if I proceed like this. Please correct me if I am wrong with my effect size expectation. For completeness I add the command pipeline used:
recon-all -s subj_name -i /.../dicom_images.dcm recon-all -s subj_name -use-gpu -mprage -all mri_convert -odt float ./... .dcm $SUBJECTS_DIR/subj_name/mri/001.mgz mris_make_surfaces -dura 001.mgz 4 subj_name lh mris_make_surfaces -dura 001.mgz 4 subj_name rh recon-all -autorecon-pial -subjid subj_name _______________________________________________ Freesurfer mailing list Freesurfer@nmr.mgh.harvard.edu https://mail.nmr.mgh.harvard.edu/mailman/listinfo/freesurfer
Hi,
I corrected my proceeding and separated the echoes using the pipeline:
recon-all -s subj_name -i /.../dicom_images.dcm recon-all -s subj_name -use-gpu -mprage -all
mri_convert -odt float -f 0 ./... .dcm $SUBJECTS_DIR/subj_name/mri/000.mgz mri_convert -odt float -f 1 ./... .dcm $SUBJECTS_DIR/subj_name/mri/001.mgz mri_convert -odt float -f 2 ./... .dcm $SUBJECTS_DIR/subj_name/mri/002.mgz mri_convert -odt float -f 3 ./... .dcm $SUBJECTS_DIR/subj_name/mri/003.mgz
mris_make_surfaces -dura %03d.mgz 4 subj_name lh mris_make_surfaces -dura %03d.mgz 4 subj_name rh
recon-all -autorecon-pial -subjid subj_name -use-gpu -mprage .
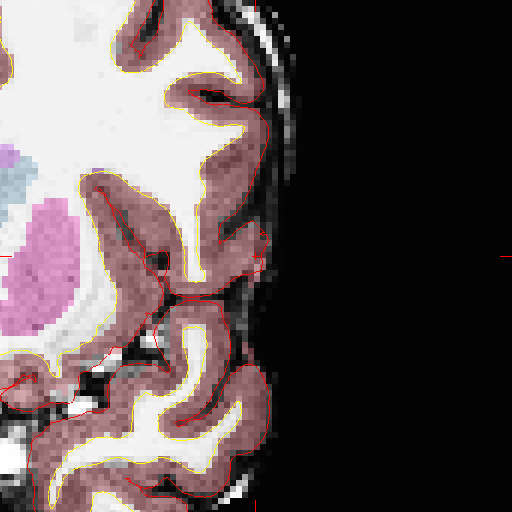
I see a difference in the cortical atlas after doing "recon-all -autorecon-pial", but if I take a look at the 3D-Volume I still find pieces of the dura included in the cortex. Please check the figure attached to this mail. The red cross signals the location.
Regards Joerg
On Wed, 12 Jun 2013 09:03:21 -0400 (EDT) Bruce Fischl fischl@nmr.mgh.harvard.edu wrote:
Hi Joerg
you need to convert the individual echoes to be in conformed space without scaling the intensities, then run it with something like
mris_make_surfaces -dura echo%d.mgz 4 subj_name lh
otherwise it will just load 001.mgz 4 times and think it is the different echoes. That is, name the echoes echo0.mgz echo1.mgz echo2.mgz and echo3.mgz
cheers Bruce
On Wed, 12 Jun 2013, Joerg Pfannmoeller wrote:
Hello FSexperts,
I used the mgh memprage and the dura correction scheme described in:
http://www.mail-archive.com/freesurfer@nmr.mgh.harvard.edu/msg28479.html .
Afterwards I applied
recon-all -autorecon-pial -subjid subj_name
to the subjects in order to include the changes in the subsequent FS-Fast analysis. Currently I do not see any effect in tksurfer if I proceed like this. Please correct me if I am wrong with my effect size expectation. For completeness I add the command pipeline used:
recon-all -s subj_name -i /.../dicom_images.dcm recon-all -s subj_name -use-gpu -mprage -all mri_convert -odt float ./... .dcm $SUBJECTS_DIR/subj_name/mri/001.mgz mris_make_surfaces -dura 001.mgz 4 subj_name lh mris_make_surfaces -dura 001.mgz 4 subj_name rh recon-all -autorecon-pial -subjid subj_name _______________________________________________ Freesurfer mailing list Freesurfer@nmr.mgh.harvard.edu https://mail.nmr.mgh.harvard.edu/mailman/listinfo/freesurfer
Freesurfer mailing list Freesurfer@nmr.mgh.harvard.edu https://mail.nmr.mgh.harvard.edu/mailman/listinfo/freesurfer
The information in this e-mail is intended only for the person to whom it is addressed. If you believe this e-mail was sent to you in error and the e-mail contains patient information, please contact the Partners Compliance HelpLine at http://www.partners.org/complianceline . If the e-mail was sent to you in error but does not contain patient information, please contact the sender and properly dispose of the e-mail.
{kind=link}
freesurfer@nmr.mgh.harvard.edu
-
 Bruce Fischl
Bruce Fischl -
 Caspar M. Schwiedrzik
Caspar M. Schwiedrzik -
 Joerg Pfannmoeller
Joerg Pfannmoeller -
Jörg Pfannmöller
-
 Welsh, Robert
Welsh, Robert